CONTROLS ON THE REDUCTION AND PRECIPITATION OF
MOLYBDENUM AND THE IMPLICATIONS FOR
GEOCHEMICAL BARRIERS
Donald J. Carpenter
Morrison Knudsen Corporation
ABSTRACT
Uncontrolled migration of elevated concentrations of uranium and its co-contaminants, including molybdenum, is a concern at numerous waste sites in the United States and around the world. One method to help attenuate and control the migration of uranium and other electrochemically reactive contaminants, such as molybdenum, is the use of a geochemical barrier. A geochemical barrier is defined as a discrete engineered layer or interval with components designed to invoke adsorption and/or precipitation of low solubility oxidized or electrochemically reduced phases. One type of geochemical barrier tailored to the precipitation of very low solubility electrochemically reduced phases of uranium and co-associated contaminants, such as molybdenum, may consist of a thin layer enriched in organic carbon. The organic carbon functions as a substrate for sulfate-reducing bacteria; these bacteria reduce dissolved sulfate ions co-associated with the dissolved uranium and other contaminants, resulting in the formation of hydrogen sulfide as a metabolic byproduct. The resulting hydrogen sulfide (or bisulfide ion) then electrochemically reduces and precipitates uranium and other co-contaminants, including molybdenum, significantly decreasing the dissolved concentration of these contaminants and immobilizing them locally. Morrison Knudsen Corporation scientists and engineers, who have pioneered in formulating this type of geochemical barrier, continue to optimize conditions that will attenuate uranium and its co-contaminants.
The effectiveness of this type of geochemical barrier is predicated on the relatively rapid electrochemical reduction and precipitation of dissolved oxidized contaminants. Any phenomenon that inhibits the rapid reduction and precipitation of the dissolved contaminants can adversely impact the effectiveness of the geochemical barrier. Certain natural systems, such as uranium sandstone-hosted "roll-front" deposits show that molybdenum is concentrated further downgradient into the pyritic and thereby "reduced" portion of the sandstone aquifer than the co-transported uranium and selenium. One model suggests this behavior is due to molybdenum requiring relatively more reducing conditions for its reduction and precipitation. An alternative explanation is that the dissolved molybdenum in this setting exists in a less electrochemically reactive form and that kinetic inhibition results in its transport relatively further into the pyritic reduced zone of the aquifer.
Determination of whether and to what extent kinetic processes contribute to the localization of molybdenum has important implications for the characteristics of a geochemical barrier. If thermodynamics alone control the localization of molybdenum, then the geochemical barrier must engender very low Eh and sulfidic conditions to ensure that molybdenum is effectively attenuated. However, if slow reduction kinetics contribute to the precipitation of dissolved molybdate, then, in addition to ensuring that a low Eh environment develops within the geochemical barrier, the barrier must also provide a significant residence and reaction time to allow for the dissolved molybdate to be slowly precipitated. If the geochemical barrier provides too little residence time, dissolved molybdenum may "escape" the engineered reducing conditions within the barrier and be released into the general environment.
This paper presents evidence that slow reaction kinetics may affect the reduction of molybdenum and, consequently, its localization in a hydrologic system. This behavior is shown to have significant consequences, especially for geochemical barriers that are not designed to accommodate this slow reduction process. Controls are presented to accommodate this behavior and effectively attenuate molybdenum, including absorption within an appropriately designed geochemical barrier.
This paper emphasizes the important of the formation of high atomic weight polymerized molybdates and their impact on the rate of electrochemical reduction. Isopolyanion development of dissolved molybdenum is shown to depend heavily on dissolved molybdenum concentration and, under acidic conditions, to a lesser extent by pH. At molybdenum concentrations greater than 10-4 molar and under acidic conditions, isopolymolybdates such as Mo7O24-6 can constitute an important fraction of the total dissolved molybdenum. These conditions are similar to those that may prevail in sulfuric acid-bearing uranium mill tailings solutions. Dissolved isopolymeric species are resistant to rapid electrochemical reduction by typical natural reductants, including sulfidic compounds. Given sufficient reaction time with sulfides, isopolymolybdates can be reduced by sulfidization, but at a slower rate than for simple mononuclear molybdates.
INTRODUCTION
The use of natural analogs may provide important insight to understanding the behavior of uranium and co-contaminants at radioactive waste sites. However, unique geochemical conditions that are rare in nature, but present at radioactive waste disposal sites can cause significant deviations from predicted geochemical behavior based on common natural analogs. This paper will present one example of how unusual geochemical conditions associated with acidic uranium mill tailings solutions may cause changes in the geochemical behavior in molybdenum from those observed in typical natural systems.
Molybdenum is known to predominately exist as a monomeric molybdate oxyanion is dilute, near-neutral pH, oxidized water (1, 2, 3, 4, 5). Monomeric molybdate anion is known though to polymerize to form high molecular weight oxyanions under elevated dissolved molybdenum concentrations and under low pH conditions (6, 7); conditions that often fall outside the conditions typically observed for many, if not indeed most, natural surface water and groundwater systems. Consequently, predicted molybdenum geochemical behavior is typically based on the predominance, or exclusive presence of monomeric molybdate ion. Whereas this presumption is usually correct, the potential for polymeric molybdate ion must be considered when non-typical low pH solutions resulting from mineral processing activities are encountered during remediation efforts. As will be shown below, at dissolved molybdenum concentrations exceeding 10-4 M and with pH conditions less than about 4.0, important amounts of dissolved polymeric molybdenum may be present at equilibrium conditions.
This paper will further discuss the conditions necessary for molybdenum polymerization, its geochemical implications, and the ramifications for engineered methods designed to help attenuate dissolved molybdenum and limit its transport at radioactive and hazardous waste sites through the use of one type of possible geochemical barrier.
CO-ASSOCIATION OF URANIUM AND MOLYBDENUM
IN NATURAL SYSTEMS
Uranium and molybdenum are closely associated in many sandstone-hosted uranium deposits, an important uranium resources (8, 9, 10, 11). Certain mill processing methods of this molybdenum enriched uranium ore result in the carryover of molybdenum into the uranium concentrates. Onward processing of this molybdenum-bearing uranium concentrate often results in a corresponding enrichment of molybdenum in the associated uranium process tailings and raffinate waste. Molybdenum is known to be toxic to various animals, particularly ruminants (3). Because of the potential for uncontrolled migration of these contaminated solutions to receptors, including susceptible animals, molybdenum is a contaminant of concern at many radioactive waste sites around the United States and elsewhere around the world.
THE CONCEPT OF URANIUM/MOLYBDENUM GEOCHEMICAL BARRIERS
One method to help attenuate and control the migration of dissolved uranium and other electrochemically reactive contaminants, such as molybdenum, is the use of a geochemical barrier. One type of geochemical barrier is an engineered system designed to invoke adsorption and/or precipitation of low solubility phases, including the precipitation of very low solubility electrochemically reduced phases of uranium, molybdenum and other electrochemically reactive contaminants. A typical geochemical barrier tailored to the precipitation of very low solubility electrochemically reduced phases of uranium and co-associated contaminants may consist of a thin layer enriched in organic carbon (12, 13). The organic carbon functions as a substrate for sulfate-reducing bacteria; these bacteria reduce dissolved sulfate ions, resulting in the formation of hydrogen sulfide as a metabolic byproduct. The resulting hydrogen sulfide (or bisulfide ion) then electrochemically reduces and precipitates oxidized uranium and other co-contaminants, including molybdenum, significantly decreasing the dissolved concentration of these contaminants and immobilizing them locally.
NATURAL ANALOGS FOR URANIUM/MOLYBDENUM
GEOCHEMICAL BARRIERS
The above type of geochemical barrier, based on the use of a reactive zone of organic carbon, is tailored to mimic the natural analog of the sandstone-hosted uranium- and molybdenum-bearing "roll-front" deposits. The formative mechanism typically postulated for these deposits involves the reaction of solubilized, oxidized species of uranium, selenium, vanadium, and molybdenum with one or more of the following reductants (8, 14, 15, 16, 17):
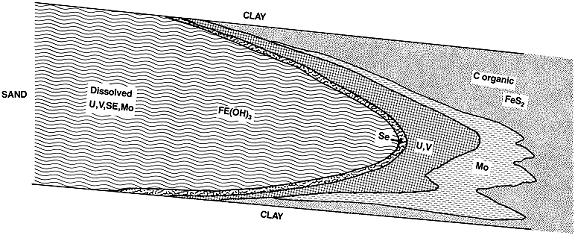
The reduction and precipitation of these electrochemically active compounds form zones enriched in one or more of the reacting elements (8, 15) as shown on Figure 1. Selenium is typically enriched in a thin band up to a few inches thick in the immediate vicinity of the oxidation-reduction boundary (the redox interface); uranium and vanadium are typically enriched immediately downgradient from the redox interface at a distance of a few feet to perhaps several tens of feet. Molybdenum is usually found even further downgradient, often up to several hundred feet from the redox interface and from the zone of maximum uranium and vanadium enrichment. The explanation for this physical displacement of the zone of maximum molybdenum enrichment from the zone of maximum uranium and vanadium enrichment is usually based solely on thermodynamics. According to this rationale, molybdenum requires the lowest electrochemically reducing (Eh) conditions for the formation of a compound that is reduced and therefore low in solubility (typically jordisite, MoS2) (8, 15). This rationale further suggests that the strongly reducing conditions necessary for molybdenum reduction and precipitation are reached only in a zone far downgradient from the redox interface. According to this view, selenium, uranium, and vanadium are precipitated under relatively less reducing conditions, closer to the redox boundary. No supporting evidence is presented for concluding that molybdenum localization is strictly caused by thermodynamic processes. Further, the typically sulfidic aquifer, containing substantial qualities of pyrite and/or marcasite and organic carbon, does not contain any "additional" reducing component downgradient in the zone of maximum molybdenum enrichment. Thermodynamic calculations, consistent with the deposition of reduced phases of molybdenum, do not indicate why molybdenum is located so far downgradient in the reduced portion of the aquifer. The above model is silent on the issue that kinetic inhibitions may inhibit molybdenum reduction and precipitation, implicitly suggesting that the reduction and precipitation of dissolved molybdenum ion is assumed to be at least commensurate with the rate of reduction and precipitation of dissolved oxidized selenium, uranium, and vanadium; an assumption that may not be necessarily correct.
Fig. 1. Reduction and precipitation of uranium and associated elements is well established in roll-front deposits.
Whereas molybdenum does require relatively lower Eh conditions to be electrochemically reduced and precipitated, an alternative explanation, or at least a possible contributing factor for the displacement of the molybdenum from the uranium, vanadium, and selenium, is the relatively slower reduction kinetics of dissolved oxidized molybdenum, a portion of which may exist in the form of high-molecular-weight isopolymolybdates.
Determination of whether and to what extent kinetic processes contribute to the localization of molybdenum has important implications for the characteristics of a geochemical barrier. If thermodynamics alone control the localization of molybdenum, then the geochemical barrier must engender very low Eh and sulfidic conditions to ensure that molybdenum is effectively attenuated. However, if slow reduction kinetics contribute to the precipitation of isopolymolybdates, then, in addition to ensuring that a low Eh environment develops within the geochemical barrier, the barrier must also provide a significant residence and reaction time to allow for the isopolymolybdates to be slowly precipitated. If the geochemical barrier provides too little residence time, dissolved molybdenum may "escape" the engineered reducing conditions within the barrier and be released into the general environment.
ISOPOLYANION FORMATION OF MOLYBDENUM
As noted above, under "typical" dilute, surface water conditions molybdenum predominately exists in a "simple" molybdate anionic form. It is important to also consider that under certain conditions molybdenum may polymerize into high molecular weight forms (6, 7). Polymerization can be first described by the reaction:
7MoO4-2 + 8H+ = Mo7O24-6 + 4H2O
Equilibria may then also be established for the following reactions:
Mo7O24-6 + H+ = HMo7O24-5
and
HMo7O24-5 + H+ = H2Mo7O24-4
Additional isopolyanionic forms of molybdenum may develop including:
Mo7O24-6 + 3H+ + HMoO4- = Mo8O26-4 + 2H2O
(Due to the absence of thermodynamic data for more high polymerized isopolymolybdates this paper will focus on forms of Mo7O24-6. Since one of the purposes of this paper is to present issues that are generally associated with the ramifications of the isopolymolybdate formation, the absence of these data do not adversely impact the overall conclusions of this paper.)
Isopolyanion development of dissolved molybdenum is shown to depend heavily on dissolved molybdenum concentration and, to a lesser extent on pH, under acidic conditions. At molybdenum concentrations greater than 10-4 molar and under acidic conditions, isopolymolybdates such as Mo7O24-6 may form a significant percentage of the total dissolved molybdenum concentration. Solution equilibria modeling, using the PHREEQC algorithm (16), demonstrates that in molybdenum-enriched, acidic solutions a significant percentage of total dissolved molybdenum can exist as isopolymolybdates. Table 1 reports the results of PHREEQC estimated equilibrium distribution of polymerized versus monomeric molybdate ions in a pH 4.0 solution with variable (and acknowledgeably elevated total molybdate concentrations). Importantly these PHREEQC results also though demonstrated that these solutions, in spite of their elevated molybdenum concentrations, were still undersaturated with respect to various oxidized molybdenum minerals.
Table 1. PHREEQC Estimated Distribution of Polymerized Versus Monomeric
Molybdate Ions - pH 4.0
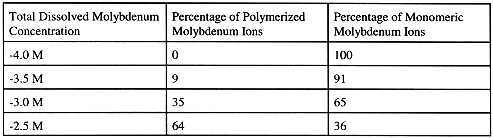
These results demonstrate that a significant percentage of total dissolved molybdenum may exist in a polymerized form and that misconceptions may result from the inappropriate extrapolation of the chemical behavior of dissolved molybdenum in dilute, near-neutral pH, surface water, where polymeric molybdenum is presumed to be absent, to the behavior of dissolved molybdenum anticipated to occur in very low-pH tailings pore water where polymeric molybdenum may be an important constituent.
THE IMPLICATIONS OF ISOPOLYMOLYBDATE FORMATION ON
MOLYBDENUM ATTENUATION
Dissolved isopolymeric species are resistant to rapid electrochemical reduction by typical natural reductants, including sulfidic compounds. Given sufficient reaction time with sulfides, isopolymolybdates can be reduced by sulfidization, but probably at a slower rate than for simple mononuclear molybdates. It is acknowledged that the actual rate is currently unknown as well as the fact that laboratory rate studies are also hampered by the generally slow reactions and the time required for establishment of equilibrium conditions (7). The sluggishness of the time necessary to reach equilibrium within the laboratory also suggests that attainment of equilibrium may be hampered in natural systems. Whereas the acidic pH conditions suitable for isopolymolybdate formation may be removed upon contact of the very acidic tailings solutions with silicate or carbonate phases present in aquifers the corresponding transformation and depolymerization of the isopolymolybdates may be delayed. Consequently, a partially neutralized solution that was derived from an initially acidic solution containing an important proportion of isopolymolybdate phases may continue to transport isopolymolybdates as metastable solutes due to the time required for depolymerization. Consequently, whenever an acidic molybdenum-enriched solution is being remediated a determination of the presence of isopolymolybdates is recommended. Further, given that isopolymolybdates are detected in important concentrations an understanding of their impact on the remedial strategy is necessary. Specifically to determine if a sufficient residence time can be provided within a geochemical barrier to allow for complete and effective reaction between the attenuating constituents of the geochemical barrier and possible refractory isopolymolybdates.
CONCLUSIONS
This paper establishes the following conclusions
REFERENCES