SURFACE AREA AND CHEMICAL REACTIVITY
CHARACTERISTICS OF URANIUM METAL
CORROSION PRODUCTS
T.C. Totemeier and R.G. Pahl
Argonne National Laboratory
P.O. Box 2528
Idaho Falls, ID 83403-2528
ABSTRACT
The results of an initial characterization of hydride-containing corrosion products from uranium metal Zero Power Physics Reactor (ZPPR) fuel plates are presented. Sorption analyses using the BET method with a krypton adsorbate were performed to measure the specific areas of corrosion product samples. The specific surface areas of the corrosion products varied from
0.66 to 1.01 m2/g. The reactivity of the products in Ar-9%O2 and Ar-20%O2 were measured at temperatures between 35° C and 150° C using a thermo-gravimetric analyzer. Ignition of the products occurred at temperatures of 150° C and above. The oxidation rates below ignition were comparable to rates observed for uranium metal.
INTRODUCTION
The movement of metallic uranium spent nuclear fuels (SNF) from long-term basin storage to interim dry storage and, for some fuels, eventual geologic repository disposal, has led to a renewed interest in the potential reactivity of uranium metal corrosion products. Corrosion products which are reactive at low temperatures in air present safety concerns during handling, drying, and dry storage of SNF which has been previously wet-stored. The main compound of interest is uranium hydride, UH3. Uranium hydride has been observed to form in the corrosion of uranium metal by water or water vapor, along with the nominal reaction product UO2. The reported percentages of UH3 in the UO2 reaction product vary from 2% to 30% [1-3]. The formation of higher percentages appears to be limited to crevice-type corrosion where there is limited access of the ambient environment to the corrosion product. UH3 forms as a fine powder and is reported to be pyrophoric at room temperature in the presence of air [4]. Burning of UH3 in air to form UO2 and H2O liberates a large amount of heat, 1,386 kJ/mol UH3. Rapid heat liberation presents a risk for contamination spread, and there is also the possibility of ignition of the underlying uranium metal.
In order to provide a better technical basis for evaluating the pyrophoric potential of corroded metallic fuels during handling and interim dry storage, the corrosion products formed during extended storage of pure uranium metal Zero Power Physics Reactor (ZPPR) fuel plates have been characterized. These plates are coupons of highly enriched uranium (HEU) metal clad in stainless steel jackets; crevice corrosion of the plates has occurred since they were originally clad in 1982. Historical experience with the corrosion products from these plates has proven them to be pyrophoric at room temperature. An extensive characterization of the ZPPR HEU plates was initiated in 1996 [5]. This characterization confirmed that the corrosion products contain large quantities of uranium hydride. A semi-quantitative X-ray diffraction (XRD) analysis estimated the weight fraction of UH3 in the loose corrosion products to be approximately 80%.
This paper presents the results of a further characterization of the corrosion products. Sorption analyses using the Brunauer, Emmet, and Teller (BET) method with Kr adsorbate were performed on the corrosion products from three different plates to measure their specific surface areas. The reactivity of the corrosion products from a single plate in Ar-9%O2 and Ar-20%O2 gas environments were measured at temperatures from 35° C to 200° C using a thermo-gravimetric analyzer (TGA). The hydride contents of the corrosion products were determined from the TGA results and XRD analysis of the reacted products.
EXPERIMENTAL PROCEDURES
Testing Apparatus
Oxidation testing of the corrosion products was carried out using a modified Shimadzu TGA-51H thermo-gravimetric analyzer (TGA). The TGA is comprised of a Cahn-type microbalance to monitor weight changes coupled with a resistance furnace to apply a temperature profile to the sample. Modifications to the Shimadzu instrument were made to allow operation of the instrument in a purified argon glovebox. The most significant changes were the connection of gas input and output lines to the TGA, and the construction of a gas control panel outside the glovebox. Both modifications were made to enable control of gas composition, flow rate, and pressure in the sample chamber. In addition, some electronic modifications were made to enable the use of a computer located outside the glovebox for TGA control and data acquisition.
The gas environment in the sample chamber is controlled by varying the rates of two separate gas flows which enter the chamber and the rate of exhaust flow out of the chamber. The first input flow is a pure Ar purge flow, which passes through the balance region of the TGA before entering the sample chamber. The second input flow contains a reaction gas, in this case a mixture of Ar and 30 vol.% O2. The reaction gas flow enters the TGA just upstream of the sample chamber, and the two flows mix prior to entry into the sample chamber. The flow rates for the two streams are set using needle valves and flowmeters in the gas control panel outside the glovebox. Pressure in the chamber is measured by a pressure transducer in the outgas line just downstream of the TGA; the pressure is controlled by varying the exhaust flow rate using a booster pump - bypass valve combination in the exhaust line.
Measurement of the specific surface areas of the corrosion products was performed on a Quantachrome QuantasorbÒ gas sorption analyzer. No modifications to the instrument were made, however, due to the radioactive nature of the samples to be tested, the analyzer was located in a contaminated fume hood. Proper operation of the instrument was verified using TiO2 and Al2O3 reference materials supplied by Quantachrome. The values measured using the Quantasorb tester agreed with the reported values (10 and 0.1 m2/g, respectively) within ± 10%.
Testing Procedures
Three ZPPR HEU plates, serial numbers 3930, 3401, and 2962, were selected to obtain corrosion products for TGA and BET testing. The plates were chosen based on their apparent degree of corrosion, evidenced by bulging of the exterior stainless steel cladding due to the volume increase of corrosion product formation. Plate 3930 had very little bulging, plate 3401 had an "average" amount of bulging, and plate 2962 had relatively severe bulging. These three plates were transferred into a pass-through Ar glovebox and declad. The atmosphere in the glovebox during the decladding procedure contained approximately 0.15% O2. The loose corrosion product from each plate was removed with a horsehair brush and examined. Contrary to indications from exterior appearance, plate 3401 contained the most loose corrosion product, 4.04 g. Plate 2962 contained 3.19 g, while plate 3930 contained 1.67 g.
The corrosion product from ZPPR fuel plates has two forms: flakes and powder. Previous XRD analysis has shown the flake material to be UO2 and the powder material to be mostly UH3 [5]. For the two more severely corroded plates (2962 and 3401), 0.01 g and 0.04 g samples of the flake-like product were removed from the corrosion products as a whole and placed into separate containers. The remainder of the products from the two plates were labeled "powder", although flake-like material was still present in the samples. After collection, the product samples from the three plates were transferred into a purified Ar glovebox for storage, loading of BET samples, and TGA testing.
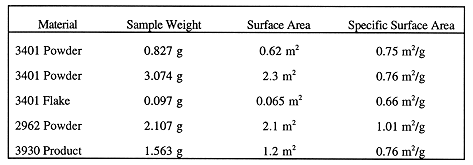
Specific surface area measurements using the BET technique were performed on samples from all three plates. The BET testing matrix and results are shown in Table I. Samples were loaded into standard sample cells in the purified Ar glovebox. The sample cells have quick-connect fittings which enable isolation of the sample from the ambient atmosphere until the cell is inserted into the analyzer for testing. Isolation of the sample prevented any reaction of the hydride with air once the cell was out of the glovebox. The loaded cells were transferred out of the glovebox to the contaminated fume hood and inserted into the analyzer. BET testing was performed using standard techniques with Kr gas as the adsorbate, He gas as the carrier, and N2 gas for calibration. Adsorption was carried out at liquid nitrogen temperature. Three consecutive measurements were made on each sample. The sample cells were transferred back into the glovebox following testing.
Table I. BET Results Summary
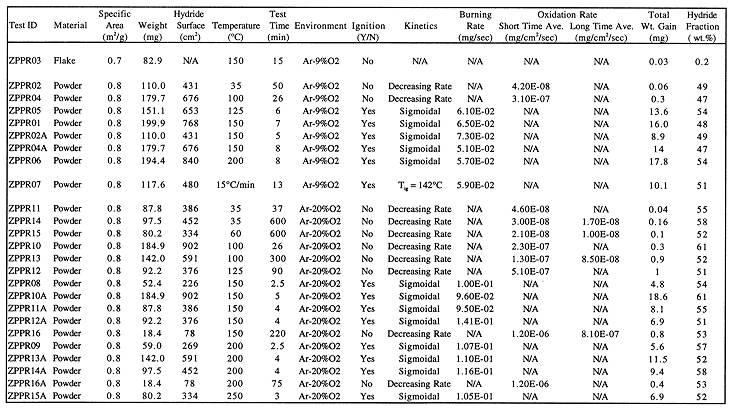
TGA oxidation testing was performed on samples from plate 3401 only. All tests but one were performed on "powder" samples from this plate. One test was performed on a flake sample. Table II shows the testing matrix and results for the TGA tests. Samples for the TGA tests were obtained from powder located in the BET sample cell by simply pouring an appropriate amount from the BET cell into the TGA sample pan. No attempts were made to ensure that the sample were representative of the bulk powder.
Table II. Oxidation Test Matrix And Results
Most tests were performed under "isothermal" conditions at temperatures ranging from 35° C to 250° C. For this type of test the TGA furnace was programmed to ramp up to the specified temperature and then maintain that temperature. During the heatup period, only pure Ar (purge gas) was flowing and present in the chamber. Once the test temperature was reached and had stabilized, oxygen was admitted into the chamber by opening the reaction gas flow line. One test performed was of the "burning curve" type. For this test, the furnace was programmed to increase the temperature at a constant rate (15° C/min) until 800° C was reached. Both purge and reacting gas flows were on during the entire heatup period. All tests were performed at a nominal pressure of 10 psi absolute, and with 200 ml/min total gas flow.
After TGA testing, all samples were visually examined for discernible changes in appearance due to oxidation. Two samples (from tests ZPPR4 and ZPPR13) were removed from the glovebox for XRD analysis. XRD was used to qualitatively determine the products of the reaction of the powder with oxygen.
RESULTS AND DISCUSSION
Specific Surface Areas of Corrosion Product Powders
Table I shows sample weights, surface areas, and specific surface areas for corrosion product samples from the three ZPPR plates. The data points shown for surface area and specific surface area represent an average of three consecutive tests on each sample. The results for "powders" were fairly consistent, varying from 0.75 to 1.01 m2/g for the three samples tested. The test of the 3401 flake material shows that its specific area (0.66 m2/g) is similar to that of the powder material. However, the small sample size (0.097 g), and hence desorption signal, reduces the confidence in this value. A second test on flake material with a much larger sample size is planned. The values obtained for both powder and flake lie within those reported in the literature for UH3, 0.3 to 0.6 m2/g by Longhurst [6], and 2.8 m2/g by Stakebake [7].
Oxidation Kinetics of Plate 3401 Product
Table II shows a summary of all TGA oxidation tests performed on samples from plate 3401, both powder and flake. The tests fall into two groups B those which showed "ignition" behavior, and those which slowly oxidized. Ignition of a sample was indicated by a high rate of weight increase and a corresponding increase in furnace control thermocouple temperature. Ignition occurred for powder samples at test temperatures of 150° C and above, with the exception of two tests discussed below.
The weight gain as a function of time during ignition and burning of samples had a sigmoidal shape. The rate of weight gain quickly increased at the beginning of the test to a fairly linear rate, then decreased near the completion of oxidation, finally dropping to zero when the sample was consumed. Burning rates corresponding to the central linear portion of the weight gain versus time curve are reported in Table II. The burning rates observed at both 9% and 20% O2 were repeatable from test to test and, more importantly, were independent of sample size or test temperature. Burning rates in Ar-9%O2 varied from 0.051 mg/sec to 0.073 mg/sec; rates in Ar-20%O2 varied from 0.095 mg/sec to 0.141 mg/sec. The lack of dependence on sample size (and hence surface area) and temperature lead to the preliminary conclusion that the burning rate was limited by mass transport in the gas phase. This assertion is corroborated by the fact that burning rates were lower for 9% oxygen concentration in the gas compared to 20% concentration.
The total weight gains of samples after completion of burning were used to calculate the weight fraction of UH3 in the sample. For tests in which ignition did not occur, a second run, denoted with the suffix "A", was run on the same sample at a higher temperature to ignite the sample and so measure the total weight gain. In order to calculate the UH3 fraction, the stoichiometry of the oxide formed in the burning reaction needed to be known. For this reason XRD analyses of reaction products from two tests were performed. Unfortunately, the results of this analysis were not conclusive. The XRD patterns showed peaks which could correspond to several different oxides, including UO2, U3O7, U3O8, and UO3. There was extensive peak overlap and broadening, which prohibited quantitative determination of the relative fractions of the various oxides. To enable a calculation of hydride fractions, all oxide produced by burning was assumed to be U3O8. This oxide had a good fit with the patterns obtained and also represents an average stoichiometry for the various possible oxides. The maximum uncertainty introduced by this assumption is 27% of the value.
The actual calculation was performed as follows. The weight gain of one gram of UH3 oxidizing to U3O8 (UO8/3) according to the reaction
|
UH3 + 4/3 O2 ® UO8/3 + 3/2 H2 |
(1) |
was calculated to be 0.168 g. The weight fraction of UH3 is then given by:
|
|
(2) |
The calculated hydride fractions for plate 3401 powder samples varied from 47 to 61 wt.%. The hydride fractions for the "powder" material do represent the bulk loose product as a whole, since the "powder" material also contained nearly all of the flake found in the plate. Of the four grams of product, only 0.1 g of flake was removed for separate testing. The remainder of the product was designated "powder". It must be noted, however, that the measured fractions do not take into account the adherent product that was not removed from the plates. Due to its adherent nature, this product is believed to be mainly oxide, and therefore the actual fraction of hydride in the corrosion product as a whole is expected to be less than the measured values for the loose product. For comparison, the fraction of hydride which can be formed during uranium corrosion by water, assuming full recycling of hydrogen, is 64.4 wt.%. Hence the observed hydride fractions are a significant fraction of the theoretical maximum for the water reaction.
The hydride fraction of each sample was used to calculate the reactive surface area of that sample. The reactive surface area was calculated as the product of the mass of the sample, the specific surface area of the material, and the fraction of hydride in the sample. The reactive surface area was used to calculate surface area normalized rate data for all tests. Because the burning rate data was independent of sample size, normalizing the burning rate data by surface area only increased the scatter in the data. Hence burning rates are reported in units of mg/sec. Obviously, the independence of burning rate on sample size is only valid for the range of sample sizes tested, 50 to 200 mg. The low-temperature oxidation rates were assumed to be surface area dependent.
The results of the one test on plate 3401 powder which was run in a burning curve mode are shown in the Fig. 1, which shows both the weight gain of the sample and the furnace control thermocouple reading as a function of test time. The control thermocouple was located approximately 2 mm below the Pt sample pan containing the sample. The sharp break in the weight gain curve at approximately 142° C clearly corresponds to the onset of ignition. There is a matching increase in the control thermocouple reading resulting from rapid heat generation in the sample. Similar increases in control thermocouple readings were also observed for isothermal tests in which ignition occurred. The post-ignition burning rate of the burning curve sample was the same as observed in isothermal tests.

Fig. 1. Plot of Weight Gain and Control Thermocouple Temperature for
Burning Curve Test.
Test ZPPR16 was the one run at 150°C in which ignition did not occur. The sample size for the test was very small, only 18.4 mg. This amount of powder was not sufficient to form a powder bed in the sample pan, unlike larger samples. The powder particles were scattered loosely on the bottom of the pan. Heat conduction away from such a sample will be much greater than for a powder bed, and it is postulated that high heat conduction raised the ignition temperature for this sample above 150°C. The second test of this sample, at 200°C (ZPPR16A), also did not show ignition.
Oxidation of plate 3401 powder at conditions for which ignition did not occur was characterized by a decreasing rate of oxidation with increasing test time. For the first several tests at lower temperature, it was thought that the rate of oxidation became linear after a short period of time, and so these tests were run only long enough to obtain what appeared to be a linear rate. Further testing, however, revealed that the rates did not become linear but continued to slowly decrease with test time. Decreasing rate kinetics are typically described as being parabolic or "paralinear" (a combination of parabolic and linear kinetics), i.e. the weight gain is dependent on t1/2 or a sum of t and t1/2 terms. Attempts to fit the low temperature data with such rate laws were unsuccessful, as was the more general approach of fitting a power law relationship to the data. Some rate laws fit the data for certain tests better than others, but no single rate law adequately described all tests.
In order to make quantitative comparisons between tests at different temperatures (and so determine an activation energy), average oxidation rates for the various tests were computed. The average rates were calculated by dividing the total weight gain at a given time by the time. Due to the decreasing oxidation rates, the average rates decreased with increasing time. For this series of tests, average rates in two time regimes were computedCa short time regime and a long time regime. For tests with durations less than 100 min, the short time average rates were computed as the weight gain at end-of-test divided by total test time. The short time average rates for tests with durations greater than 100 min were computed as the weight gain at 100 min divided by 100 min. Similarly, the long time average rates for tests with durations between 100 min and 300 min were computed as the weight gain at end-of-test divided by total test time, while the rates for tests longer than 300 min were computed as the weight gain at 300 min divided by 300 min. As mentioned above, the oxidation rates for all low-temperature tests were surface area normalized.
The rate of oxidation below the ignition temperature generally increased with increasing testing temperature. Figure 2 is a summary plot showing weight gain as a function of time for longer-duration tests in Ar-20%O2. With the exception of the 35° C and 60° C tests, the rate of oxidation increases with increasing temperature. It is believed that that rates of oxidation at the two lowest temperatures may have been sufficiently low that normal drift in the TGA baseline reading over time may have obscured the real weight gain.
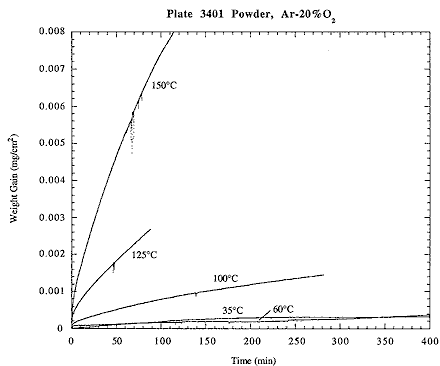
Fig. 2. Plot of Weight Gain Versus Time for Longer-duration Tests in Ar-20%O2.
Figure 3 is an Arrhenius plot of the low-temperature oxidation rates. The short time average rates for Ar-9%O2 and Ar-20%O2 are shown along with the long time average rates for Ar-20%O2. There is considerable scatter in the data; the data for the different time averages and oxygen concentrations are not significantly different in comparison with this scatter. A straight line fit to the log rate versus inverse temperature plot yields the following rate equation for oxidation of plate 3401 powder in Ar-9%O2 and Ar-20%O2:
|
k = 2x10-2 exp [B36,000/RT] mg/cm2/sec |
(3) |
where k is a linear rate constant, R is given in units of J/K/mol, and T is the temperature in Kelvin.

Fig. 3. Low-temperature Oxidation Rates Versus Reciprocal Temperature.
The results from the test performed on the flake sample from plate 3401 (ZPPR7) showed that there was no significant quantity of hydride associated with this sample. The weight change over a 15 min period at 150° C was 0.01 mg. All powder samples showed very significant weight changesCeither burning or fairly rapid oxidationCover a similar time period. The flake sample was visually unchanged after testing. The stability of the flake material was also evidenced by its behavior in powder tests. Because not all flake was separated from the plate 3401 corrosion product, some flake material was present in the nominally "powder" samples. This flake remained visually unchanged following oxidation or ignition of the sample, unlike the powder, which experienced an increase in volume due to oxidation.
The data obtained in this series of tests compares somewhat favorably with the limited amount of information available in the literature. Both Longhurst [6] and Stakebake [7] have performed oxidation tests with uranium hydride. Longhurst reported that little or no reaction occurred at room temperature (possibly due to oxide impurities), but observed ignition at 140° C, which is very close to the value of 142° C obtained in this series of tests. The rates reported by Stakebake at temperatures between 0° C and 100° C appear to match well with the burning rates observed for plate 3401 powder, approximately 0.1 mg/sec of weight gain. The temperatures at which these rates occurred were much lower than in this series of tests, indicating that ignition occurred at lower temperatures. The lower ignition temperature could be due to a higher specific surface area (2.8 m2/g) for Stakebake's hydride material, or less oxygen contamination present on the hydride.
The low temperature oxidation rates are comparable to oxidation rates for uranium metal in dry air published in a review by Ritchie [8]. Figure 4 shows the rates for uranium metal and those for the current tests. One apparent difference in the data is a lower activation energy (slope) for uranium hydride oxidation compared to uranium metal oxidation. The reason for this difference is unknown.

Fig. 4. Comparison with U Oxidation Data from Ritchie [8].
CONCLUSIONS
Initial testing of the corrosion products found on uranium metal ZPPR fuel plates was performed. Characterization of the products included specific surface area determination and measurement of oxidation kinetics in Ar-20%O2 and Ar-9%O2. The following conclusions were reached:
|
k = 2x10-2 exp [B36,000/RT] |
(3) |
where R is in units of J/K/mol and T is the absolute temperature in Kelvin.
ACKNOWLEDGMENTS
The authors would like to acknowledge D.M. Pace, R.W. Bratt, and E.A. Beverly with assistance in setting up the TGA and BET analyzers, and S.M. Frank for performing the XRD analysis. This work was supported by the U.S. Department of Energy National Spent Nuclear Fuel Program.
REFERENCES