INTEGRATED DIRECT CHEMICAL OXIDATION SYSTEM FOR
THE NON-THERMAL DESTRUCTION OF CHLOROSOLVENTS
Bryan Balazs, John Cooper, Peter Hsu, Pat Lewis, Joe Penland
Larry Finnie, Martyn Adamson
Chemistry and Materials Science Directorate
Lawrence Livermore National Laboratory
Livermore, CA 94550
ABSTRACT
An integrated bench-scale system for the non-thermal destruction of a wide range of chlorinated organic liquids and organic-contaminated sludges has been demonstrated at our facility at LLNL. Chemical process data have been developed for this system, e.g., for the destruction of materials such as 1,1,1-trichloroethane, trichloroethylene, perchloroethylene, and on the treatment of surrogate sludges contaminated with model chloro-organics. This aqueous based system operates at near ambient temperature and employs two separate and distinct processes for the mineralization of chloro-organics to carbon dioxide and chlorine. Many chlorinated organic liquids, such as solvents, present difficulties in their destruction due to their high volatility and limited solubility in water, and the described system circumvents these problems. The first step of the process is a hydrolysis of the chlorosolvents, performed at elevated temperatures in a sealed reactor with minimal headspace. Following this step, the resultant products, which are generally more soluble and less volatile, are mineralized in a process known as Direct Chemical Oxidation (DCO). The DCO technology employs aqueous solutions of peroxydisulfate at 80-100 oC to oxidize hydrolysis products to carbon dioxide and chlorine. If desired, the expended oxidant can be recycled electrochemically to minimize secondary waste. Because of the physical and chemical operating parameters of this system, it is also applicable to the treatment of mixed waste.
INTRODUCTION
The destruction of chlorinated waste through traditional thermal techniques often encounters regulatory or political hurdles, primarily when there is concern about the formation of dioxins or furans. When the waste stream under consideration is mixed waste, the problem is compounded due to possible volatilization or other mechanisms for the release of radionuclides into the environment. For these reasons, alternatives to traditional thermal organic waste treatment technologies are being considered, and there are several techniques under various stages of development. [1]
Direct Chemical Oxidation
One of these alternative techniques developed over the past five years at LLNL is known as Direct Chemical Oxidation (DCO). [2-10] DCO is a non-thermal, ambient pressure, aqueous based technology for the oxidative destruction of the organic components of hazardous or mixed waste streams. The process uses solutions of peroxydisulfate salts (typically sodium or ammonium) to completely mineralize the organics to carbon dioxide and water, and the expended oxidant may be electrolytically regenerated to minimize secondary waste. The net waste treatment reaction is (Eq. 1):
S2O82- + (organics) Þ 2HSO4- + (CO2, H2O, inorganic residues) |
(1) |
Peroxydisulfate is one of the strongest oxidants known (oxidation potential is +2.05V), and is exceeded only in oxidative power by fluorine, ozone, and oxyfluorides. The oxidation potential of peroxydisulfate is high enough to oxidize nearly all organics, and thus the process is virtually "omnivorous." Results by this laboratory and others show that the intrinsic rate constants for the oxidation of a variety of organics all lie within a factor of two of a median value, i.e., 0.02 min.-1. [11-16]
Mild thermal activation (80-100 oC) of peroxydisulfate solutions results in the generation of the sulfate free radical, which is the active oxidizing agent (Eq. 2):
S2O82- Þ 2SO4- |
(2) |
Alternatively, this radical-generation process may be effected at near ambient temperatures (30-50 oC) through the use of a catalyst such as platinum, silver or copper. The reaction of the sulfate free radial with the organic and with water results in a cascade of active oxidants including organic free radical fragments, hydroxyl free radicals, peroxymonosulfate (a strong industrial bleach), hydrogen peroxide, etc.
The ammonium (or sodium) hydrogen sulfate can be recycled in a flowing electrolyte cell, using a platinum or glassy carbon anode, a graphite or oxygen-depolarized cathode, and a porous ceramic separator to prevent cathodic reduction of the product. The anodic reaction is (Eq. 3):
2NH4HSO4 Þ (NH4)2S2O8 + 2H+ + 2 e- (anode) |
(3) |
while the cathodic reaction is the reduction of protons, or of water itself, to form hydrogen gas.
Volatilization Loss with Chlorosolvents
The operating regime of the DCO system is typically 80-100 oC, which is often well above the boiling point of chlorinated solvents such as 1,1,1-trichloroethane, methylene chloride, trichloroethylene, etc. As a result, these materials are often swept away into the offgas of a DCO reactor and there is not enough time of contact with the oxidant solution for destruction to commence. Results from our laboratory showed that with organic substrates listed above, typical values for conversion to carbon dioxide were 50% or less, with no organic remaining in the oxidant solution at the end of a run. Offgas analysis showed that the organic substrate volatilized before quantitative oxidation could occur. Varieties of reactor design and operating parameters proved ineffective at remediating this problem.
Hydrolysis as a Pre-step to DCO
Neutral and acid hydrolysis of many chlorinated organics is known to occur under conditions of ambient pressure and temperature (17-20). In many cases, the hydrolysis product is a more soluble and less volatile material as in the example of hydrolysis of 1,1,1-trichloroethane (Eq. 4)
CH3CCl3 + 2H2O Þ CH3COOH + 3H+ + 3Cl- |
(4) |
However, the rates of these reactions are often measured in terms of years at ambient temperatures. By raising the temperature of the hydrolysis reactor, and operating in a sealed system with minimal headspace, we have shown that the hydrolysis reaction can completed in a matter of hours. Subsequent oxidation of the hydrolysis products via the nominal DCO parameters results in quantitative conversion of the original chlorinated organic to carbon dioxide and chlorine. This process is applicable on either liquid organics, or contaminated soils/sludges. We describe here a bench scale, integrated system as a demonstration of this more versatile technology for chlorosolvent destruction.
EXPERIMENTAL
All chemicals were reagent grade; all water was distilled. Sodium hydroxide was used for the hydrolysis reactions while sodium peroxydisulfate was used during oxidation reactions.
Laboratory Scale System
Laboratory scale hydrolysis reactions were done in a stainless steel (304) pressure vessel with an internal immersion heater, with an internal volume of 2.1 liters. Mixing was done by using an oversize (3-4") stir bar driven by an external magnetic stir plate. An electronic pressure transducer and overpressure relief valve were fitted. All components in contact with the internal fluid were either Teflon coated, or stainless steel (304 or 316).
Laboratory scale oxidation reactions were done in a one liter glass vessel with magnetic stirring, fitted with a reflux condenser and external heating. The offgas from this reactor was drawn through a trap consisting of a potassium iodide solution (in this manner the chlorine released could be determined by iodimetry) and then to an infra-red carbon dioxide analyzer. The value for the carbon dioxide concentration in the offgas was, along with the instantaneous flow rate, recorded using a data acquisition interface with LabView; the two values could thus be used to quantitatively determine an integrated volume of carbon dioxide released from the destruction of the organic.
Integrated Bench Scale System
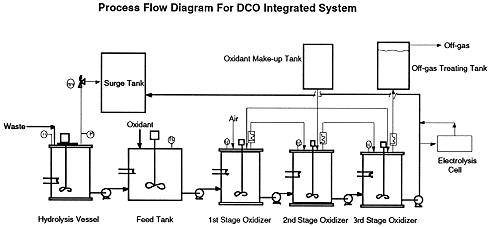
Figure 1 shows the integrated bench scale system which includes a hydrolysis vessel, a feed tank, a 3-stage continuous stirred tank reactors (CSTR), an off-gas treatment tank, an oxidant make-up tank, as well as a surge tank. An electrolysis cell can be included in the process as secondary equipment for the regeneration of oxidant. The hydrolysis vessel was made of 316L stainless steel with an internal volume of 73.2 liters. This vessel was equipped with a 5kW immersion heater and a double mechanical seal mixer with a seal rated at 200 psig and 150 oC. This vessel was designed for a maximum operating pressure of 100 psig and maximum operating temperature of 120 oC. An electronic pressure transducer was fitted and a pressure relief valve was fitted and set to 120 psig. Fluids were pumped in and out of the vessel using appropriate valving and peristaltic pumps. The organic substrate was pumped in first, followed by the appropriate quantity of sodium hydroxide solution. Following a hydrolysis run, the hydrolyzed solution was then transferred to a 30 gallon polypropylene feed tank where oxidant is added at room temperature. Excess oxidant is needed for more favorable kinetics and throughput. The solution is then cascaded through the oxidation system.
Fig. 1. Schematic of Integrated Direct Chemical Oxidation System
The oxidation system consisted of three CSTR's. These reactors were glass and of an internal volume of 17 liters. The flow rate and residence time in the CSTR can be carefully controlled such that desirable destruction removal efficiencies (DRE’s) can be achieved. Samples are taken from each stage for analyses of TOC and oxidant. The 3rd stage CSTR is used as a polisher where oxidant concentration and operating temperature vary in order to completely destroy all the organics in the solution. The organic-free solution can then be discharged into a 55 gallon surge tank or passed to an electrolysis cell for the regeneration of oxidant to reduce cost and minimize the oxidant consumption. The offgas from each CSTR was pumped through a condenser, and then each output was combined into one common stream. A slipstream of this was passed to the carbon dioxide analyzer setup used in the laboratory scale experiments above, while the flow rates of the slipstream and of the main stream were measured continuously. Liquid samples for analysis were taken from each CSTR output as needed.
RESULTS AND CONCLUSIONS
Hydrolysis Conditions
Although the literature references (17-20) indicate that the hydrolysis of many chlorinated solvents is through a reaction with water, and not necessarily hydroxide ions, basic conditions were chosen for the present work. The reason for this is related to the integrity of the hydrolysis tank. During the hydrolysis reaction, protons as well as chloride atoms are released and this would lead to much more rapid attack on the stainless steel material of construction. Since this tank operates at pressure and temperature, it was believed that for safety reasons, it was more prudent to insure that the pH of the hydrolysis vessel contents was always maintained at a high value where the corrosion would be less severe. Indeed, when the amount of hydroxide was intentionally made insufficient, resulting in acidic conditions (pH <2) at the end of the experiment, XRF analysis of the hydrolysis liquid showed high concentrations of stainless steel components, such as Fe, Cr, Ni, etc. If the quantity of base is sufficient, protons released from reaction (4) would immediately react with hydroxide and thus be precluded from existing in high concentrations. The amount of base necessary to insure this result was calculated (assuming a sufficient safety factor) using equations such as (4) and assuming complete hydrolysis of the quantity of chloro-organic used. It was determined in laboratory scale experiments that the hydrolysis reaction proceeded well under highly basic (pH > 14.0) conditions and that a pH of at least 13.5 was maintained throughout the experiment.
Laboratory Scale Hydrolysis Reactions
A number of hydrolysis experiments were run at the laboratory scale to determine the rates of hydrolysis, and the reaction conditions, and the results are shown in Table I. The extent of hydrolysis was measured by measuring the amount of insoluble, second phase organic material left over at the end. While this method is not extremely accurate and does not identify products specifically, it was deemed sufficient as the aim of the hydrolysis step is to produce a single liquid phase for subsequent oxidation. The rate of oxidation of organics by peroxydisulfate is very high at 80-100 oC, being essentially mass transport limited, and providing a single aqueous phase is a major step towards a successful system.
Table I. Hydrolysis and oxidation data for laboratory scale system
| Chlorosolvent(s)* | Time, Temp. of Hydrolysis |
% Hydrolyzed |
% Conv. to CO2 |
| 1% 1,1,1-trichloroethane | 5 hr @ 100 oC |
100 |
100 |
| 3% 1,1,1-trichloroethane | 4 hr @ 120 oC |
100 |
90 |
| 10% 1,1,1-trichloroethane | 6 hr @ 110 oC |
100 |
- |
| 2% carbon tetrachloride | 6 hr @ 112 oC |
25 |
8 |
| 0.6% each of: 1,1,1-trichloroethane, perchloroethylene trichloroethylene methylene chloride chloroform |
6 hr @ 120 oC |
92 |
78 |
| 1% each of: perchloroethylene trichloroethylene |
5 hr @ 115 oC |
75 |
40 |
* The numbers refer to the volume percent loading of chlorosolvent in the hydrolysis vessel.
A GC-MS analysis of the hydrolysis product liquid showed that the hydrolysis of 1,1,1-trichloroethane did not lead to a single product as would be indicated by Eq. (4). Although acetic acid (or acetate ion under basic conditions) was a major product, other products were chloromethane, methylene chloride, chloroform, carbon tetrachloride and 1,1,2-trichloroethane. In addition, a dark brown solid material formed during hydrolysis which, although soluble in organic solvents, was not soluble in water. Generally, the quantity was less than about 10 vol% of the initial chlorosolvent substrate. Through IR and GC-MS analysis, it was determined that this material was a complex mix of larger chloro-organics including 1,2,3-trichlorobenzene, trichloronaphthalene, trichloropentene, tetrachloropentane and C10H8Cl8 (Mol. Wt. 408).
It thus appears that the hydrolysis reaction involves not only the breakage of C-Cl bonds, but also breakage of C-C bonds and subsequent rearrangements. This result, while unexpected, is not necessarily detrimental as these materials are formed in relatively small quantities, and would be expected to be easily destroyed by peroxydisulfate in the subsequent oxidation step. As an example, the fact that many of these compounds are unsaturated or aromatic is actually an advantage in the reaction with peroxydisulfate.
In general, the hydrolysis was quantitative for a variety of organic substrates, with the notable exception of carbon tetrachloride. This material hydrolyzed only very slowly, and it is estimated that a hydrolysis reaction at the temperatures used would require weeks. Of course, increasing the temperature would decrease the time requirement, but this was not possible with our system. For all chlorinated organics, it was very important that the mixing of the hydrolysis solution be well designed, as early runs gave poor performance due to erratic mixing. The importance of proper mixing is discussed in more detail in the Integrated System discussion below.
Laboratory Scale Oxidation Reactions
The products of the hydrolysis reactions for a variety of chlorinated organics were subsequently reacted with peroxydisulfate under the nominal DCO conditions, and the extent of conversion to carbon dioxide was measured (see Table I) Note that in some cases the oxidation was done under the basic hydrolysis conditions, while in others the solution was acidified using sulfuric acid before the oxidation step. There are two important differences between these two pH regimes. First, in basic media, carbonate is the product of oxidation and remains in solution until the pH drops below about 6, at which point carbon dioxide is rapidly evolved in a single sharp peak (see Figure 2). The drop in pH from basic to acidic is due to the reduction of peroxydisulfate by the organic, with bisulfate as the product (Eq. 1). In acidic media, the oxidation evolved carbon dioxide almost immediately after reaching approximately 70 oC (see Figure 2). The second difference is that in acidic media, the organic chloride is released as chlorine, presumably through the direct recombination of chloride free radicals. In base, no chlorine gas is evolved, at least until the solution pH falls to values of 1-2 (again, the pH drop is due to the reduction of peroxydisulfate as mentioned above).
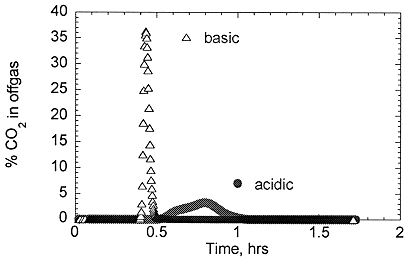
Fig. 2. Evolution of CO2 during the oxidation of 1,1,1-trichloroethane hydrolysis product
In general, the conversion of the organic to carbon dioxide was
quantitative, at least within the accuracy of the described measurement technique. When
the results indicated less than 100% conversion to carbon dioxide, it was believed to be
due to the fact that the hydrolysis product, i.e., the oxidation feed stream, contained
solid particles which were not always transferred properly into the oxidation reactor.
Total Organic Carbon analyses done on the solution after oxidation always indicated
essentially negligible carbon content, at least to the detection limit of the TOC analyzer
(» 1ppm carbon).
Integrated System Results
The above data at the laboratory scale were used to design and build the Integrated System described in the Experimental Section above and shown in Figure 1. This system has been tested on 1,1,1-trichloroethane, at both 3 and 10 vol% loading, and on a mixture of chlorosolvents. The parameters for the completed runs are given in Table II.
Table II. Summary of Experimental Runs for Integrated System
Run # |
Organic Substrate(s)1 |
Time, Temp. of Hydrolysis |
DRE |
1 |
test (ethylene glycol) |
not applicable |
>90% |
2 |
test (acetic acid) |
not applicable |
>97% |
3 |
test (acetic acid) |
not applicable |
>99% |
4 |
3% 1,1,1-trichloroethane |
4 hr @ 120 oC2 |
>98% |
5 |
3% 1,1,1-trichloroethane |
4 hr @ 120 oC2 |
>99.5% |
6 |
3% 1,1,1-trichloroethane |
2.5 hr @ 110 oC |
>99.5% |
7 |
mixture of: |
2.5 hr @ 110 oC |
see text |
8 |
10% 1,1,1-trichloroethane |
4 hr @ 110 oC |
see text |
1
The numbers refer to the volume percent loading of chlorosolvent in the hydrolysis vessel.Mixing the reaction fluid in each DCO reactor is easily achieved since it is a single homogeneous phase. Mixing speed was maintained at above 100 rpm to obtain turbulent flow in order to reduce mass transfer resistance and improve the process kinetics. The required mixing time is fairly short (<20 seconds) for incoming liquids comparing to the residence time which was kept at above 50 minutes. For mixing the two-phase liquids in the hydrolysis vessel, a properly designed impeller and high mixing speed are required to minimize mass transfer resistance and increase interfacial surface area for faster hydrolysis. The mixing was achieved with a 5 inch high shear impeller (Shearmor impeller by MixMor) at 800 rpm. The combination of powerful mixing and a high reaction temperature significantly reduced the time required for complete hydrolysis from years to several hours.
As the hydrolysis of the volatile chlorosolvents proceeds, the pressure inside the hydrolysis tank will decrease assuming that the products or intermediates formed are less volatile than the starting material. For hydrolysis reactions of materials such as 1,1,1-trichloroethane, this was expected to be the case as acetic acid is a primary product of the reaction. This was verified experimentally as shown in Figure 3, where the pressure and temperature of the hydrolysis vessel are plotted versus time. For the first 3 hours, both pressure and temperature increase as the reactor is heating up. There are slight increases in pressure for 3 or more hours, and then the pressure falls off rapidly. The increase in pressure for the intermediates when the temperature is relatively constant is presumably due to the formation of more volatile intermediates.
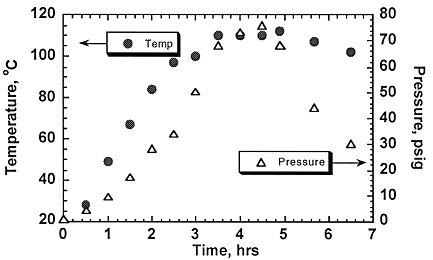
Fig. 3. Pressure and temperature profile for hydrolysis of 1,1,1-trichloroethane
With this integrated system, several different chlorosolvents or chlorosolvent mixtures were tested as shown in Table II. In the earlier runs #4 and #5, the hydrolysis was done using multiple batches of the smaller, laboratory scale mixer, which were then combined for the oxidation runs. The Destruction and Removal Efficiency (DRE) was measured by measuring the amount of organic or inorganic carbon remaining in the waste discharge from the system. These listed DRE values are the results averaged over a run; instantaneous values were often higher (>99.9%). Due to the limitations of the analysis method used, the limit of detection of carbon corresponds to a DRE of about 99.9%, and thus some of the instantaneous values may be even higher. Note that oxidation of the hydrolysis output by the trio of CSTR’s was not done in all cases (runs 7 and 8), as this portion of the system had already been proven and additional runs would simple result in excess waste with little new data.
The results correspond well with the theoretical process model predictions, based on kinetic constants. A more detailed set of data for a run with 1,1,1-trichloroethane is shown in Table III. In each of the three CSTR’s, the average DRE matches the predicted value almost exactly. It appears that the relationship is slightly better than expected for the first CSTR, while being slightly less than expected for the third. This is no doubt due to the decreasing oxidant concentration in the third reactor, and a simple remedy would be to add an oxidant makeup system to this final stage as a final polishing step.
Table III. Process Model vs. Actual Data for Integrated System Run #6
Time, hr |
DRE, CSTR#1 |
DRE, CSTR#2 |
DRE, CSTR#3 |
0.00 |
94.64% |
99.18% |
- |
0.58 |
96.82% |
99.77% |
99.81% |
1.17 |
95.36% |
99.58% |
99.86% |
2.00 |
92.73% |
99.32% |
99.65% |
2.33 |
92.73% |
- |
99.70% |
Average |
94.45% |
99.46% |
99.76% |
Theory |
93.63% |
99.59% |
99.97% |
CONCLUSIONS
We have demonstrated that an Integrated System consisting of a hydrolysis at moderate temperature, followed by Direct Chemical Oxidation using the oxidant peroxydisulfate, can be used to quantitatively destroy a number of volatile and heavily chlorinated organic solvents. The hydrolysis of these solvents results in less volatile, more soluble intermediates, thus enabling the oxidation step to proceed with minimal loss of organic substrate in the offgas. We believe that this process should be applicable to many other chlorinated solvents in addition to the ones tested.
Our group at Lawrence Livermore is current engaged in a treatability study to destroy 300 kg of low-level, mixed chlorosolvents currently stored at LLNL. These mixed chlorosolvents contain primarily 1,1,1-trichloroethane, perchloroethylene, trichloroethylene, with a number of other materials in trace amounts. The reactor system designed and built for this treatability study is conceptually identical to the one described here, and actual productions runs are scheduled for early FY98.
This work was performed under the auspices of the U.S. Department of Energy by the Lawrence Livermore National Laboratory under contract number W-7405-ENG-48.
REFERENCES
BACK