WET CHEMICAL OXIDATION AND STABILIZATION OF MIXED
AND LOW LEVEL ORGANIC WASTES
R. A. Pierce, R. R. Livingston, and D. A. Burge
Westinghouse Savannah River Company
P.O. Box 616
Aiken, SC 29802
W. G. Ramsey
CeraChem Technologies
227 Gateway Drive
Aiken, SC 29803
ABSTRACT
Mixed acid oxidation is a non-incineration process capable of destroying organic compounds, including papers, plastics, resins, and oils, at moderate temperatures and pressures. The technology, developed at the Savannah River Site, uses a mixture of an oxidant (nitric acid) and a carrier acid (phosphoric acid). The carrier acid acts as a holding medium which allows appreciable amounts of the oxidant to be retained in solution at atmospheric pressure and at the temperatures needed for oxidation. The phosphoric acid also provides the raw materials for making a final waste which contains the metal contaminants from the waste stream.
Savannah River has designed, built, and started up a 40-liter pilot reaction vessel to demonstrate the process and its sub-systems on a larger scale than earlier testing. The unit has been demonstrated and has provided important data on the operation of the oxidation and acid recovery systems. Specific results will be presented on oxidation conditions, acid recovery efficiency, chloride removal, metal retention, and process monitoring.
Additional studies have been conducted with a smaller vessel in a radioactive hood. Testing with plutonium-bearing waste simulants was performed to make preliminary predictions about the behavior of plutonium in the process. Samples of the remaining phosphoric acid from these tests has been converted to two separate final forms for analysis. Results will be presented on plutonium fractionation during the oxidation process and waste form stability.
INTRODUCTION
The purpose of this program is to demonstrate a nitric-phosphoric acid destruction technology which can treat a heterogeneous waste by oxidizing the solid and liquid organic compounds while decontaminating noncombustible items. The process will operate below 200oC and at atmospheric pressure for most materials and moderate pressures (< 250 kPa) for complex organics, and will convert hazardous organics and organic substrates to gases and inorganic salts. This development will produce a complete, closed-loop, engineering-scale process which produces little or no organic residue and isolates hazardous and radioactive metals from solution as an iron phosphate glass or phosphate-based ceramic. Of particular interest to the customer is the treatment of solid, TRU-contaminated job-control waste (a heterogeneous mixture of plastics, cellulose, lead, rubber, resins, solvents, oils, steel, ceramics, HEPA filters, etc.). The technology proposed is unique to the Savannah River Site (SRS).
The basis for the process stems from extensive studies conducted at WSRC.1,2,3 The process contains three distinct parts: organic oxidation, acid recycle, and metal stabilization. The oxidation step uses HNO3 in a concentrated phosphoric acid media as the main oxidant for the organic compounds. The nitric acid products from the oxidation, NO2 and NO, can be regenerated in an acid recovery system using air and hydrogen peroxide. Other oxidation byproducts, such as HCl (a byproduct of PVC oxidation) must removed from the offgas stream. The oxidation of the waste to gaseous products leaves all hazardous ions in solution. Once oxidation is complete, the phosphoric acid stream becomes the primary feedstream for immobilizing the hazardous ions in either an iron phosphate glass (1050-1150oC)4 or a magnesium phosphate ceramic (room temperature).5 Materials that have been oxidized include neoprene, cellulose, nitromethane, polyethylene, PVC, divinylbenzene resins, and oils.
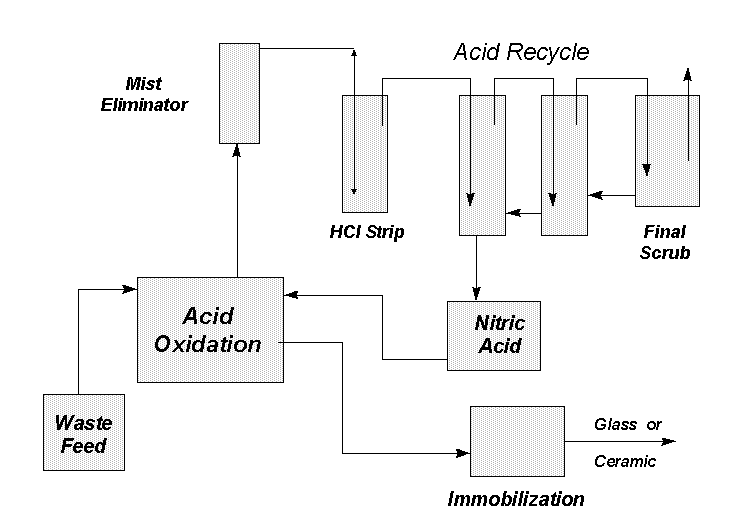
Fig. 1. General Flowsheet
EXPERIMENTAL DISCUSSION AND RESULTS
Development Areas
Many different areas were studied and evaluated over the past year. All of the efforts focused on developing a system which could be scaled up to a pilot-scale process. The areas studied include construction of an engineering-scale unit, oxidation at the bench and engineering scale, HCl removal, NOx recovery and recycle, process monitoring, retention of plutonium and uranium, and final waste forms.
Construction of an Engineering-Scale Unit
Discussion: Many things have been demonstrated at the bench scale, but often many operability issues surface when scaling a system up to the production scale. Therefore, it is essential to conduct pilot-scale testing of the oxidation and nitric acid recycle systems. Attention will be placed on the performance of the acid recycle system. Specific testing was performed to measure radionuclide retention, chloride partitioning, NOx recycle, and hydrogen peroxide use efficiency. Additional consideration will be given to process monitoring and will allow us to evaluate the hardware and software selections made in constructing this 40-liter system so as to provide feedback for the next scale up.
Results: Information is provided here to give some background into the design. The 40-liter pilot-scale vessel was made from glass with a diameter of 0.3m and a height of 0.6m. Glass was chosen due to uncertainties associated with other materials of construction. Although glass has a relatively high corrosion rate (> 1.0 mm/yr), it was judged to be resistant enough while providing a material which allows a viewing of the experiments. Due to the fact that glass was used, operating pressures were limited to 205 kPa. The oxidation vessel was also fitted with a glass stirrer and a heating mantle; the heating mantle was controlled using a PID temperature controller. A mist eliminator was also manufactured for the offgas stream to limit metals carryover.
Energy balance calculations were performed for the oxidation and acid recycle processes to determine if chillers would be needed for the pilot-scale system. Calculations indicated that a maximum of 6.6 kW of cooling capacity would be needed for the oxidation vessel, 1.9 kW for the condenser, and 3.1 kW for the acid recycle system. A tantalum cooling coil supplied with process water was built for the oxidation vessel while closed-loop chillers were used for the condenser and acid recycle system.
Acid recycle vessels were fabricated in the SRTC Glass Shop to allow visualization of the experiments. The oxidation vessel heater was ordered as a two-circuit heating mantle with 2.4kW of heating capacity. Cooling coils for the acid recycle system were designed and fabricated using 304-L stainless steel. Type J thermocouples were used for all temperature measurements, pressure transducers for pressure measurements, and rotameters for gas flow measurements. Temperature and pressure readings were continuously monitored using Opto 22 hardware and Wonderware software. The use of pumps was not a major concern based on prior experience in the lab-scale systems. All pumps selected for the pilot were variable-speed peristaltic pumps. All pumps were set up to use viton flexible tubing which has exceptional chemical resistance.
Oxidation Testing
Discussion: A key to determining the viability of the process is to determine the effectiveness of oxidation and oxidation rates for the target waste stream and other applicable streams. Also, it is necessary to begin to assess the cumulative effect of continuous oxidation on the oxidation process. Compounds which were studied include cellulose, neoprene, polyethylene, and PVC. The objective of the experiments was not to obtain the maximum possible rate, but rather what is understood to be the maximum practicable rate based on desired temperatures and pressures.
Results: The earliest tests aimed at determining the amount of cellulosic material which can be dissolved into a fixed volume of phosphoric acid. This helps estimate expected volume reductions during overall processing. When complete, 240 grams of KimWipesTM were oxidized in 70 mL of phosphoric acid. Some precipitates started to form after 120 grams and the oxidation rate was being impeded by the presence of other ions in solution. Examination of the solids clearly indicates that they are silicates from cellulose; plastics should not have these impurities. The residual phosphoric acid was then immobilized into 43 mL of iron phosphate glass without difficulty. This is a waste loading density of 5600kg/m3. Based on estimates from SRS Solid Waste Management, 210 liter waste drums have approximately 11.5kg of organic waste, or 54.5kg/m3. Even if it is assumed that all organic in a drum is cellulose, volume reduction should be on the order of 50-100X depending on where the processing endpoint is established.
Another test was run to show the oxidation profile which represents the release of carbon dioxide gas from the destruction of a waste mixture containing paper, polyethylene, neoprene, and PVC. Three batch additions were made an hour apart and oxidation was observed at 200oC and 185-205 kPa. As expected, paper dissolved within three minutes, polyethylene within 15 minutes, neoprene within 30 minutes, and PVC at about an hour. Although neoprene has a faster oxidation rate than polyethylene, its thickness and resistance to melting cause it to take longer to dissolve.
A series of engineering-scale experiments were conducted in the 40-liter vessel with approximately 16.5 liters of phosphoric acid. Tests included the oxidation of individual compounds (cellulose, neoprene, and polyethylene [PE]) at a range of parameters such as temperature, pressure, and acid concentration. A table of batch testing results are shown below. These reflect what has been observed during lab-scale tests. It is clear that there are temperature, pressure, and acid concentration effects as has been reported earlier.
Table 1. Engineering - Scale Batch Tests
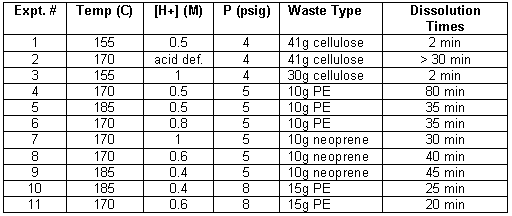
Batch tests were also conducted with a mixture of cellulose, neoprene, polyethylene, and PVC at 170-185oC, 0.5-1.0M HNO3, and 155-170 kPa. Consistent with the results above, oxidation of cellulose, neoprene, and polyethylene at 170oC and 1.0M HNO3 occurred at similar rates as at 185oC and 0.5M HNO3; in both cases the neoprene and polyethylene were dissolved inside of 15 minutes. It is expected that the accelerated rate is caused by the dissolution of cellulose which increases the NOx concentration in the solution
Difficulties were encountered with feeding solids in a continuous manner. The net result is that the engineering-scale unit could not be tested in continuous mode. Consequently, follow-up work was performed at bench-scale to evaluate throughput capabilities for different organics.
Tests were run is a vessel containing 200 mL of phosphoric acid in a semi-continuous mode. Samples were added every 15 minutes over a two-hour period. Polyethylene bag was run in which 1.0 gram samples are added each 15 minutes at 185oC and 170-185 kPa. The samples dissolve readily throughout the experiment and are oxidized. The rate of addition corresponds to a throughput rate of 206 kg C per 24 hours per cubic meter vessel (assuming 50% freeboard). When the experiment is repeated using a thicker, higher density polyethylene (from bottles). Overall dissolution to completion is slower because of the increase in thickness and density; materials take 50 minutes to dissolve instead of 10-15 minutes. However, the continuous dissolution rate eventually stabilizes and appears to be consistent with that of the polyethylene bag. Testing with neoprene had not yet been initiated at the end of the fiscal year.
Because of the apparent resistance of PVC to dissolution at 185oC and 170 kPa, oxidation testing was conducted using mixtures of organics in the feed. Tests were run with mixtures of cellulose, neoprene, polyethylene, and PVC. The oxidation characteristics of these tests appear to be consistent with lab-scale experience which show PVC to be the most resistant to oxidation. Based on observations, it is believed that PVC dissolution rate, even when PVC is added in small quantities, will be the rate limiting step in the process. As a result, tests were run at temperatures just above the melting point of PVC (202oC). These showed oxidation of PVC at a rate of approximately 35-40 g/liter-hr. However, the raising of the reaction temperature would mean lower concentrations of nitric acid remain soluble, or that system pressure would need to be increased. A separate experiment showed that at 205oC and 205 kPa, 0.24M of nitric acid is soluble. At 220oC this falls to 0.02M while at 190oC it is 0.60M.
Chloride Removal
Discussion: The need to remove chloride from the system exists because of byproducts from the oxidation of PVC and neoprene. When oxidized, the chloride forms HCl which is sufficiently volatile to escape into the offgas system. If not removed separately, the HCl will end up in the acid recycle system and, subsequently, build up in the system. The most expedient method for eliminating HCl involves using water in a bubbler system to absorb the HCl.
Theoretical calculations based on chemical equations in the literature for warm water solutions suggest that nitric acid concentrations in a water wash would build up to a maximum level long before the HCl would reach a maximum. Beyond the maximum absorption, NOx would pass through the chloride wash bottle and be absorbed in the first hydrogen peroxide acid recovery vessel. It is expected that if the concentration of nitric acid in the chloride wash were to get too high, chloride would be able to pass through the system as either HCl or NOCl.
Results: Pilot testing showed that the chloride wash bottle effectively removed chloride from the offgas from 0-2000 ppm. Data also showed no detectable chloride remaining in the oxidation pot or passing through to the first hydrogen peroxide bottle. However, at the same time it was observed that essentially all of the NOx was also being absorbed in the chloride wash bottle from 0-2.8M nitric acid. This was considered potentially problematic. As a result, a small scale experiment observed the chloride and nitric acid scrubbing effects in the chloride wash bottle over a wider range of concentrations. The results, contrary to theory the proposed, indicate that both NOx and HCl absorb until they reach a "collective" maximum (Figure 2). What this means is that there would be no point where HCl would absorb while NOx remained unabsorbed, a condition which is unacceptable for acid recycle.
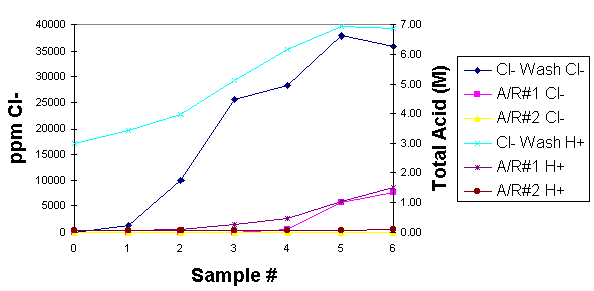
Fig. 2. Theoretical HNO3 - HCL Absorption
Follow-up testing at the bench scale evaluated a simple condenser for the removal of HCl and water vapor without significant absorption of NOx. The first experiment using a simple, small-volume condenser showed retention of approximately 50% of the HCl while allowing for NOx passage through the system. The condenser arrangement was then modified to allow for larger condenser volumes to improve condensation of both HCl and water. The modified arrangement yielded slightly better results at 57%. It is unclear at this time whether a condenser needs to be made which has higher contact area for condensing HCl or whether the lack of HCl retention stems from the formation of NOCl caused by the reaction of NOx and available chloride.
NOx Recovery and Recycle
Discussion: Several approaches exist for the recycle of nitric acid. Some options are proprietary technology of which little is known aside from vendor claims. The most attractive alternative for radioactive service is hydrogen peroxide absorption. Calculations of the maximum theoretical nitric acid concentration expected for absorption of NO2 using hydrogen peroxide indicates that high nitric acid concentrations could be obtained using 30% hydrogen peroxide. Using the equation
2 NO2 + H2O2 - - - - - > 2 HNO3 |
(1) |
it was determined that a maximum concentration of 61.3 wt% could be obtained with 30% H2O2 and 78.7 wt% with 50% H2O2. These calculations do not account for any acid formation which may occur due to NO2 absorption by the balance of water in the hydrogen peroxide solution.
It is important to note that the presence of NO gas in the stream reduces the maximum theoretical concentration. The reaction of NO with H2O2 is as follows:
2 NO + 3 H2O2 - - - - - > 2 HNO3 + 2 H2O |
(2) |
Not only is peroxide use efficiency reduced, but there is also a dilution effect from the water in the reaction products. This yields theoretical maximum acid concentrations of 47.0 wt% for 30% H2O2 and 60.8 wt% for 50% H2O2.
Results: Experiments were conducted to study NOx absorption into hydrogen peroxide to confirm design parameters for the pilot-scale acid recycle unit. Four key parameters were evaluated: NO:O2 ratio, residence time to convert NO to NO2 prior to absorption, percent hydrogen peroxide, and use of oxygen versus air to convert NO to NO2. A series of twelve tests were run in which NO was generated, pumped at a constant rate, mixed with an oxygen source fed at a constant rate, through hydrogen peroxide. The liquid was then titrated to determine acid concentrations. Some of the results are shown in Figure 3.
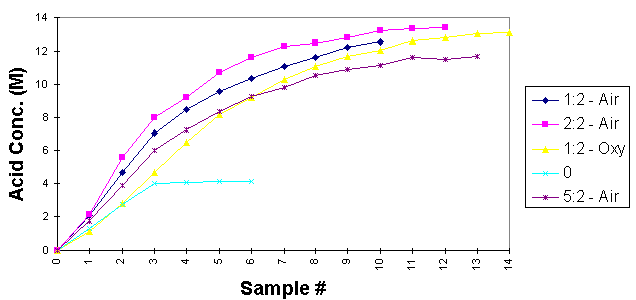
Fig. 3. Absorption in 30% Peroxide (RT= 60 sec)
The most important things to note from the data are as follows: 1) tests with pure NO yield a nitric acid concentration of 4.1M compared to NO and air which yields approximately 13M; 2) controlled absorption conditions have consistently shown conversion of 30% hydrogen peroxide to above 13M which is well above the 11.5M measured under less controlled conditions in FY96; 3) 50% hydrogen peroxide does not seem to offer an advantage over 30% peroxide commensurate with the higher cost, concentration, and chemical instability; 4) oxygen does not seem to provide any significant benefit over air; 5) the optimum residence time prior to NO2 absorption is on the order of 60 seconds; the optimum NO:O2 ratio is 2:2 but ratios as low as 1:2 reduce NOx absorption less than 10%.
Experiments were also run using three hydrogen peroxide bottles in series and flowing a small amount of NOx through the system to show how the NOx distributes through the three bottles as a function of acid concentration in the peroxide. The test showed that the hydrogen peroxide in the first bottle will absorb NOx completely until it is about two-thirds consumed before allowing NOx to pass through to the second bottle.
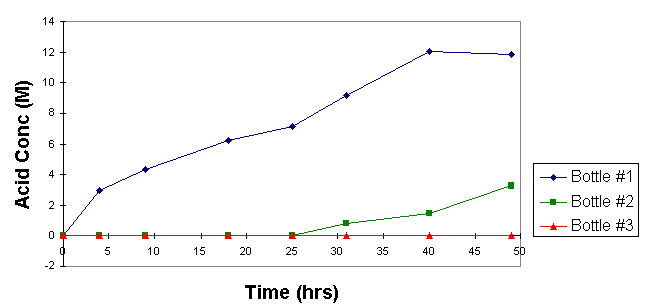
Fig. 4. Sequential NOx Absorption
Also, throughout the test, continuous monitoring of NOx emissions from the third peroxide bottle always showed 30 ppm or less as compared to the clean air standard limit of 200 ppm. Absorption efficiency is a function of geometry, but is indicative of what can be expected during the proper use of hydrogen peroxide units in series to absorb NOx to recycle nitric acid.
Process Monitoring
Discussion: For safety, process control, and optimization reasons, it is necessary to be able to monitor and control the process using reliable instrumentation on a near-continuous basis. While many of the process readings can be taken using standard thermocouples and pressure transducers, the monitoring of nitric acid in the oxidation vessel is clearly an area in need of technology development. The use of spectrometry is being strongly considered and evaluated against other analytical methods for this application. Material compatibility issues and the need for real-time information preclude the use of some "standard" analytical methods.
Results: Monitoring nitric acid concentration for the system has required the innovative assembly of components to allow real-time, at-line, analysis of the process. The requirements established for this analysis required analytical results at 10 second intervals over a concentration range of 0.1 to 1.5M nitric acid in the presence of concentrated phosphoric acid. The accuracy desired was 0.05M over the analytical range with a precision of 0.02M in order to track acid consumption for real-time control.
Early in the process design the use of a miniature fiber-optic spectrometer was envisioned in order to combine monitoring capability for iron and plutonium in addition to the nitric acid concentration. This system was anticipated to operate at 300 nm for the nitrate measurement; however, our early tests demonstrated multiple interferences at this wavelength. The alternative was to make the measurements at 210 -240 nm range. The nitrate absorbance at the lower wavelength is about 1000 times more sensitive, allowing application of a shorter flowcell pathlength and minimizing of interference.
The potential of placing a probe into the reaction vessel was explored for a brief time and abandoned based on current state of technology and our limited development budget. As test of interfering compounds continued, we discovered that high concentrations of iron impacted the nitrate concentration measurement. The mechanism for this effect has not been determined; however, after diluting the sample solution 1:10 results agreed with calculated values. By using a dilution on the process solutions we are able to minimize the corrosion of stainless steel analyzer components and aid in cooling the solution. Dilution over a range of 1:10 to 1:50 were made and provide sufficient analyzer accuracy and precision. In practice, the minimum dilution and pathlength are used to minimize sample dilution water which will be returned to the process. There is additional need to study the effect of metal nitrates formed during this process.
The system is calibrated and calibration checks are performed by pulling sample from standard solutions instead of the process vessel. The spectrophotometer operating software automatically applies a calculation model to sample measurements and provides a near real-time readout of the process nitrate concentration. Further software development will be required to allow this number to be used for feedback control. This software is also capable of using multiple models over various wavelength ranges so the visible absorption of iron and plutonium can be measured simultaneously.
Monitoring of additional process streams using a single spectrophotometer takes advantage of multiplexing technology and is an added benefit of using fiber-optic equipped spectrophotometers. UV absorption analyzers have been demonstrated by others to measure NO, NO2, and H2O2 for process control. With this in mind, we can anticipate using the nitrate monitoring instrument to monitor off-gas along with nitric acid build-up and peroxide depletion in the acid recovery unit.
Metal Retention/Behavior of Plutonium and Uranium
Discussion: When organic materials contaminated with radioactive metals are oxidized, the radioactive metals are given a certain "mobility" within the system. It is expected that the metals will dissolve into the mixed acid bath and remain there until it is time to immobilize the spent acid. It is important to retain the radioactive metals in the mixed acid in order to prevent contamination from entering the acid recovery system and escaping the overall process through the offgas system. Extra benefits can be drawn if radioactivity in the acid recovery system can be kept sufficiently low so as to permit the placement of vessels outside of containment or in areas of increased operator access.
Results: Plutonium experiments were conducted to understand how much carryover of plutonium, if any, occurs during operation. A series of tests were conducted using paper, neoprene, and polyethylene at varying process conditions and system air flows. In our experiments, a phosphoric acid solution containing 1X106 dpm/mL of Pu-239 was made and used to contaminate samples and as the oxidation vessel liquid. The system has a 1.5 liter oxidation vessel containing 600 mL of phosphoric acid. Nitric acid is pumped into the system as needed. Gases leaving the system flow through a column of glass beads (which functions as a mist eliminator) before passing through a series of three wash bottles representing the acid recovery system. Analytical data for a series 10 tests with small quantities of paper, neoprene and polyethylene showed that the alpha activity in the three wash bottles for all three test conditions was below the detection limit of 6.5E+01 dpm/mL for all samples.
Next, a set of experiments was conducted with increasing amounts of cellulose added as a batch ranging from one gram to five grams to examine what happens during high reaction rates and large volumes of gas generation.. Although gas generation rates were extremely high, alpha activity in the wash bottles was below the detection limit.
One longer test processed 25 grams of cellulose through 600 mL of solution over one hour. Consistent with previous results, no carryover of plutonium was detected in the wash bottles. A total organic carbon analysis of the first wash bottle from this run showed only 395 ppm of organic carbon in 100 mL of hydrogen peroxide. A rinse of the mist eliminator using 20 mL of dilute acid showed 6.7E+02 dpm/mL of alpha activity had accumulated for the entire set of 17 experiments.
A final test was run with six cellulose filters, each weighing about 3 grams, containing a total of 2.5 grams of uranium oxide powder between them. Consistent with the plutonium data, uranium was below the 0.01 ppm detection limit in the wash bottles. A 10 mL rinse of the mist eliminator detected 0.088 ppm of uranium. The final uranium content of the phosphoric acid was 3600 ppm.
Final Waste Forms
Discussion: The value of any organic waste treatment process is strongly dependent upon the ability to safely and conveniently place any hazardous or radioactive metals in a stable final waste form. The use of a phosphoric acid matrix provides at least two final waste forms, iron phosphate (FeP) glass and magnesium phosphate (MgP) ceramic. In comparison, the ceramic is more convenient to make while the glass provides a greater volume reduction and more stable form. The ceramic form was studied because sufficient data already exists on the stability of iron phosphate glass.
Results: Work was conducted with Argonne National Laboratory (ANL), the primary developers of magnesium phosphate ceramics for waste immobilization. Past work at ANL has shown the ceramic to be sufficiently stable to pass TCLP. Samples of magnesium phosphate ceramic were made with an oxidation process waste acid stream contaminated with both plutonium and uranium.
Two different compositions were made and no problems were experienced as the acid was effectively and easily immobilized. The first sample used a composition proposed by ANL. In the test, 30 grams of 90% H3PO4, 29.7g H2O, 19.5 g KOH, 13.8 g MgO, and 10.6 g of KH2PO4. We started with the H3PO4 and then gradually added a mixture of the KOH and water over a 20-25 minute period. Next, a mixture of the MgO and KH2PO4 were added all at once. The solution was stirred for seven minutes and then poured into a mold. The solution was stirred in the mold for an extra two minutes before being allowed to cure over the next 30-45 minutes. The sample cured for two weeks before being submitted for leach testing.
In the second sample, the water content was reduced by 4.2 grams. Due to the reduced water content, the second sample got much hotter and began to set up quicker. The sample was stirred for one minute (as compared to seven minutes in sample 1) before being poured in the mold. The sample was stirred an additional three to four minutes before it began solidify to an extent that stirring need to stop. The sample was allowed to cure for two weeks before submitting for leach testing.
The samples were leach tested using a modified TCLP method. The results show that Si, PO43- and NO3- leach from the second sample at 2-3 times the rates observed in the first sample. Furthermore, the first sample showed less that 0.007 mg/L Mg leached versus 309 mg/L Mg for the second sample. Discussions with ANL indicated that the variation is linked with the rapid solidification of the second sample which inhibited proper curing. Due to problems in the analysis, samples had to be re-submitted for analysis of potassium and uranium.
CONCLUSIONS
Experimental work in advanced the technology towards demonstrating viability as a production-scale system. Key issues such as NOx recycle, materials of construction, dioxin formation, process monitoring, final waste form development, radionuclide entrainment, and operability were successfully addressed. Concerns regarding waste throughput were also addressed on a small scale, but were limited due to waste feeding problems at the 40-liter scale. Increases can be realized through elevated temperatures and pressures well within specified limits, but are not desired by the current customer. Open issues still exist with regards to chloride stripping, nitric acid volatility, and waste feeding – solutions have been proposed, but have not yet been evaluated.
REFERENCES
BACK