THE SEPARATION AND DETERMINATION OF PLUTONIUM IN TWO LOW
LEVEL RADIOACTIVE WASTE STREAMS
K.I. Burns and D.W. Everall
AECL
Chemistry and
Chemical Engineering Division
Chalk River Laboratories
Chalk River,
Ontario, Canada K0J 1J0
ABSTRACT
A program was initiated at Chalk River Laboratories (CRL) to determine the
physical, chemical and radiological properties of wastes intended for disposal
in IRUS (Intrusion Resistant Underground Structure),
a belowground vault to be constructed at CRL. The isotopes of plutonium are
among the most restrictive radionuclides for IRUS due to their high
radiotoxicity and long half lives. A radiochemical method has been developed to
determine 238Pu, 239+240Pu and 242Pu in two
waste streams, incinerator ash and liquid feed to a bitumenizer. Samples are
spiked with 236Pu tracer, dried and fused at 960°C with Li2B4O7/LiBO2
(2:1) in a platinum boat. The plutonium is then separated by solvent extraction,
electrodeposited onto stainless steel counting planchettes and measured by
 -spectrometry. Limits of
detection for plutonium in solids are typically 0.01 Bq/g based on a 1-gram
sample, a 4-hour counting period, and a 5% counting efficiency. This paper
presents a summary of the method and the results from analysis of the two waste
streams.
-spectrometry. Limits of
detection for plutonium in solids are typically 0.01 Bq/g based on a 1-gram
sample, a 4-hour counting period, and a 5% counting efficiency. This paper
presents a summary of the method and the results from analysis of the two waste
streams.
INTRODUCTION
In 1982, in support of its new waste disposal program, Chalk River
Laboratories (CRL) initiated a Waste Categorization and Routing Program (WC&RP)
to determine the physical, chemical and radiological properties of wastes. The
near-term objective for the WC&RP was to develop the technologies and
establish the procedures for characterizing candidate wastes destined for
disposal in IRUS (Intrusion Resistant Underground Structure),
a belowground disposal vault to be constructed at CRL. These radioactive wastes
have been accumulated from 1946 to the present at CRL from on-site activities
and off-site waste producers (universities, hospitals, etc.). Some information
on the chemical and radionuclide inventory of these wastes was known but it was
recognized that more information would be required to support a disposal case.
Two waste streams that were selected for initial characterization by the WC&RP
were incinerator ash from the CRL radioactive waste incinerator, and active
drain waste concentrated through reverse osmosis prior to its solidification in
bitumen. These wastes were selected for initial characterization by the WC&RP
for the following reasons: 1) these wastes appeared to be IRUS candidate wastes;
2) they represented a significant fraction of the CRL operational wastes; 3)
they contained mixtures offission and activation products representative of the
types of wastes generated across CRL; 4) the wastes contained low levels of
these radionuclides which permitted easy handling and sampling for analysis; and
5) the radiochemical methods developed for these wastes might be easily adapted
for analysis of other wastes.
As with most disposal facilities, the inventories of the long-lived
- and
 -emitting radionuclides are
highly restricted because they do not decay appreciably during the lifetime of
the facility. One of the more restrictive radioelements for IRUS is plutonium.
An analytical method has been developed to determine the
-emitting isotopes of
plutonium in both waste streams, based on solvent extraction followed by
electroplating and measurement by
-spectrometry.
-emitting radionuclides are
highly restricted because they do not decay appreciably during the lifetime of
the facility. One of the more restrictive radioelements for IRUS is plutonium.
An analytical method has been developed to determine the
-emitting isotopes of
plutonium in both waste streams, based on solvent extraction followed by
electroplating and measurement by
-spectrometry.
This paper presents a summary of the method and the results for the two
waste streams. The results have been used in modeling the performance of IRUS to
demonstrate that these wastes can be safely disposed in the facility.
RADIOCHEMICAL ANALYSIS PROGRAM
The radiochemical analysis program was initiated in 1982 to determine the
long-lived non- -emitting
radionuclides in CRL candidate waste streams for IRUS. The objectives of the
radiochemical analysis program were to: 1) develop the radioanalytical methods
capable of determining these radionuclides in the various waste forms destined
for disposal; and 2) attempt to establish correlation factors between the -emitting and non--emitting radionuclides to
enable the latter to be estimated non-destructively through the use of -ray spectrometry. The
criteria for the radiochemical methods were that they should: 1) have adequate
sensitivity to measure the activity concentration of a radionuclide in the
waste, typically down to 0.01 to 1 Bq/g; 2) yield radiochemically pure
radionuclides for measurement; and 3) be easily adapted for analysis of a
variety of waste streams.
-emitting
radionuclides in CRL candidate waste streams for IRUS. The objectives of the
radiochemical analysis program were to: 1) develop the radioanalytical methods
capable of determining these radionuclides in the various waste forms destined
for disposal; and 2) attempt to establish correlation factors between the -emitting and non--emitting radionuclides to
enable the latter to be estimated non-destructively through the use of -ray spectrometry. The
criteria for the radiochemical methods were that they should: 1) have adequate
sensitivity to measure the activity concentration of a radionuclide in the
waste, typically down to 0.01 to 1 Bq/g; 2) yield radiochemically pure
radionuclides for measurement; and 3) be easily adapted for analysis of a
variety of waste streams.
EXPERIMENTAL
Sampling and Analysis of Two Waste Streams
Radioactive ash, which is no longer produced, resulted from operation of the
CRL incinerator during the period 1982 to 1989. The ash was initially stored in
approximately 200 drums (each of a volume of 210L), however, it was finally
bitumenized to improve its stability and reduce the volume. Of the 200 drums of
ash, 80 were assayed for -emitting radionuclides
with a -monitor
and 14 were selected for sampling and destructive analysis prior to
bituminization based on the presence of high levels of fission and activation
products, or on unusual distributions of these two types of radionuclides. The
procedure used for sampling the 14 drums, and homogenizing the ash samples, has
been described previously (1). The samples were checked for homogeneity by
measuring the radionuclide activity concentrations in 1.0-, 5.0- and 10.0-g
samples from each sample bottle by -ray spectrometry. If the
-ray
spectrometry indicated that the sample was homogeneous, a 0.5- to 1.0-g
sub-sample was taken for destructive radiochemical analysis for plutonium as
described in the procedure below.
Liquid feed to the bitumenizer originates as effluent from the CRL active
drain system, and is stored in a holding tank from where it is fed to the Waste
Treatment Center (AC). It is filtered and concentrated through reverse osmosis
and evaporation in the AC, prior to being mixed and solidified with bitumen in
0.21-m3 drums. Each bitumen campaign generates approximately 3 to 5
of these 0.21-m3 drums of bitumenized waste which have been
concentrated from approximately 120,000 to 200,000 liters of active drain waste.
Prior to bituminization, a sample containing approximately 1 L of the liquid
feed to the bitumenizer is taken by operations staff at the WTC after vortex
mixing of the liquid feed, and 500 mL are sent to the laboratory for -spectrometry and
radiochemical analysis. The samples typically consist of two phases (supernate
and solid precipitate), which are thoroughly mixed by shaking for one minute,
prior to sampling for -
spectrometry and destructive analysis. For -spectrometry, a weighed
aliquot (10 to 25mL) is filtered, and the -emitting radionuclide
activity concentrations in the liquid and solid fractions are determined
separately. The -emitting
radionuclide activities in the two fractions are then added together to
determine the overall activity concentrations in the liquid feed. This approach
is necessary to eliminate any bias that would be introduced from settling of the
suspended solids during the measurement by -spectrometry. A weighed
aliquot (0.5 to 1.0 mL) of the feed solution is taken at the same time for
destructive radiochemical analysis for plutonium as described in the procedure
below.
Plutonium Sample Dissolution
Direct acid attack was not successful in completely dissolving ash or liquid
feed samples due to the presence of silica, insoluble refractory oxides (Al2O3,
ZrO2), and insoluble salts (eg. CaF2). Experience at
this laboratory in destructively analyzing a wide variety of solids for various
radionuclides has demonstrated that high temperature fusions are the preferred
method to ensure both complete dissolution of the matrix and isotopic exchange
between an added tracer and the desired radioelement. Even after complete
dissolution, plutonium can undergo numerous hydrolytic and radiolytic reactions
which produce complex ions or polymeric colloids or cause changes in oxidation
state with time. The formation of these species with different oxidation states
can often result in low recoveries of plutonium. Our experience has shown that
fusion with a 2:1 mixture of lithium tetraborate to lithium metaborate (Li2B4O7:LiBO2),
followed by dissolution in 1.5 mole/L HNO3, yields a clear solution
from which plutonium can be extracted using tri-octyl phosphine oxide (TOPO)
without further sample treatment. Extractions performed within a reasonable time
(one week) on samples treated in this way typically result in overall recoveries
of 80 to 95% after electroplating. Approximately 0.2 to 1.5 Bq of 236Pu
tracer (AEA Technology, AEA Fuel Services Division, Process Services Division,
B220 Harwell Laboratory, Oxfordshire, OX11 ORA) are added directly to a weighed
sample aliquot in a platinum crucible. The sample is then dried in an oven at
120°C for one hour, and ashed at 500°C for 30 minutes. A 4:1 weight
ratio of the Li2B4O7/LiBO2 flux
to sample is added to the crucible and the mixture fused at 960°C for one
hour. The sample is finally cooled and dissolved in approximately 100 mL of 1.5
mole/L HNO3. Once dissolved, solutions are stable for a period of
one week. If the samples contain high concentrations (5 to 10w/o) of the
elements calcium, aluminum and silicon, precipitates (calcium and aluminum
borosilicates) may form after prolonged storage (one to two weeks) which can
adsorb plutonium and result in low recoveries.
Plutonium Separation
Plutonium separation on a sample should be carried out within a week of
dissolution of the flux, otherwise the sample should be evaporated to near
dryness several times with concentrated HNO3 containing 1% HF to convert all the
plutonium to Pu (IV). Table I lists the
-emitting radionuclides
that cannot be resolved from the various plutonium isotopes using
-spectrometry, and
therefore must be removed during the radiochemical separations. The two
radiochemical separation procedures for plutonium outlined below remove these
interferences.
Table I Interfering Radionuclides in Plutonium Measurement by
-Spectrometry

If the sample aliquot is known to contain less than 5 µg of uranium,
the sample is made to contain 2 to 8 mole/L HNO3 by the addition of
concentrated HNO3 and the plutonium is extracted into an equal
volume of 0.1 mole/L TOPO in toluene (2). Both Pu (IV) and Pu (VI), along with
uranium, thorium and neptunium (IV), are extracted under these conditions (3).
The plutonium is back extracted into an equal volume of 0.1mole/L ammonium
bioxalate from which it is electroplated onto a stainless steel counting
planchette for measurement by -spectrometry. Although thorium and uranium follow
the plutonium through the procedure, as long as they are less than 5 µg in
the sample aliquot, they do not degrade the plutonium
-spectrum significantly.
For those samples that contain more than 5 µg of uranium or that
require higher decontamination factors, the plutonium is first extracted using
2-the noyltrifluoroacetone (TTA) (4,5). In this case, the sample is adjusted to
contain 1 mole/L HNO3 and the plutonium is extracted into an equal
volume of 0.5 mole/L TTA in toluene. The TTA layer is washed with 1 mole/L HNO3
and the plutonium is back extracted into an equal volume of 8 mole/L HNO3.
The TTA layer containing the uranium is discarded, and the plutonium in the
aqueous layer is extracted into an equal volume of 0.1 mole/L TOPO in toluene.
The TOPO layer is washed with 0.1 mole/L HNO3, and the plutonium is
finally back extracted into an equal volume of 0.1 mole/L ammonium bioxalate
from which the plutonium is electroplated. Using this procedure to determine
plutonium in low enrichment uranium, decontamination factors of 107
from uranium have been achieved.
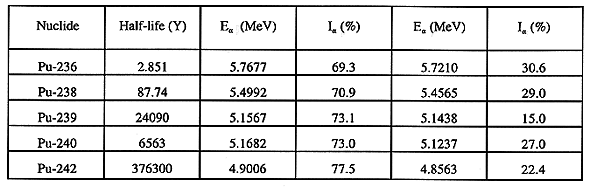
The activity concentrations of 238Pu, 239+240Pu and
242Pu are determined from the intensities of their 5.4992,
5.1567+5.1682, and 4.9006 MeV
-particle emission energies
respectively, corrected for detector efficiency, yield from the 236Pu
tracer, and normalized to sample weight. The nuclear data used in the
calculation of the various activity concentrations of the plutonium isotopes is
summarized in Table II.
Table II Nuclear Data for Plutonium Isotopes
Electrodeposition and Alpha Spectrometry
In order to produce the highest quality sources for measurement by
-spectrometry, while at the
same time ensuring reproducible source geometries, this laboratory employs
electroplating for final source preparation. A diagram of the electroplating
cells, which were constructed in this laboratory, is presented in Fig.1. The
power supplies used for electroplating were simple and inexpensive AC adapters
which supply 12V DC at 1000 mA current.
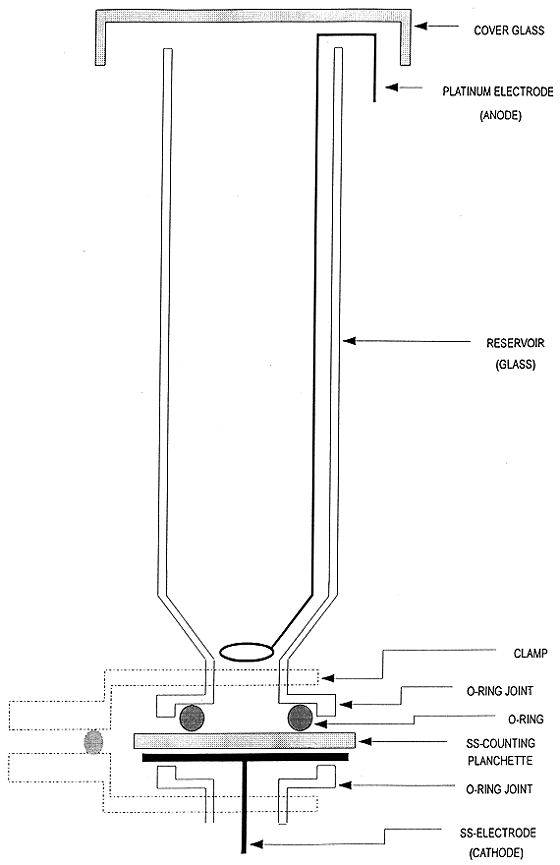
Fig. 1. Electroplating cell.
The sample, in 0.1 M ammonium bioxalate, is transferred to the cell along
with 1.0 mL of 5.0 mole/L NH4Cl as the electrolyte (6). The power is
turned on and the sample is allowed to electroplate for a period of 8 to 16
hours to ensure at least 60% of the plutonium is electroplated. Prior to turning
off the power, 1.0mL of 7 mole/L NH4OH is added to make the solution
basic to prevent any redissolution of the plated plutonium (6). The power is
turned off, the solution discarded, the cell disassembled and the planchette
removed. The planchette is rinsed first with water, followed by isopropyl
alcohol and allowed to air dry. Plutonium sources prepared in this manner
typically have a resolution (full width at half maximum) of 20 to 35 keV. A
typical -spectrum of
plutonium isotopes separated and electroplated according to the above procedures
is presented in Fig. 2.
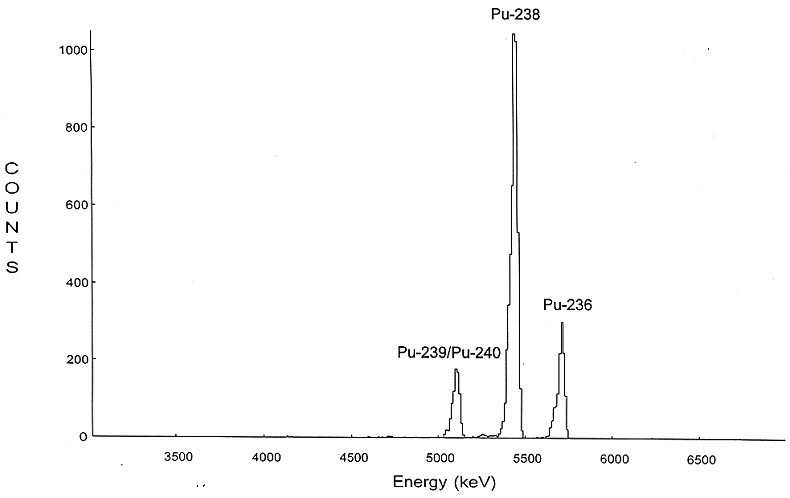
Fig. 2. Example of spectrum of Pu
fraction following TOPO extraction and electrodeposition.
Plutonium measurements were made on an eight-channel Canberra Alpha Analyst
spectroscopy system equipped with Canberra model A-450-20-AM alpha PIPS
detectors. The Analyst is interfaced to a personal computer (model P5-100
Pentium) for system control, data storage and processing using Genie PC-based
alpha analysis software. The system was calibrated for efficiency response as a
function of -particle energy using an Amersham electroplated standard alpha
source 9734RA containing 241Am,239Pu and 244Cm.
RESULTS AND DISCUSSION
The activity concentrations of plutonium and a number of
-emitting radionuclides
have been determined in 14 samples of ash and 30 samples of liquid feed to the
bitumenizer. The range in activity concentrations of the various radionuclides
measured in ash and liquid feed to the bitumenizer has been summarized in Table
III. These concentrations were used together with the estimated volume of each
waste stream going into IRUS, to calculate the total inventory of each
radionuclide in the first IRUS unit.
Table III Range in Activity Concentrations of Radionuclides in Ash,
and Liquid Feed to the Bitumenizer
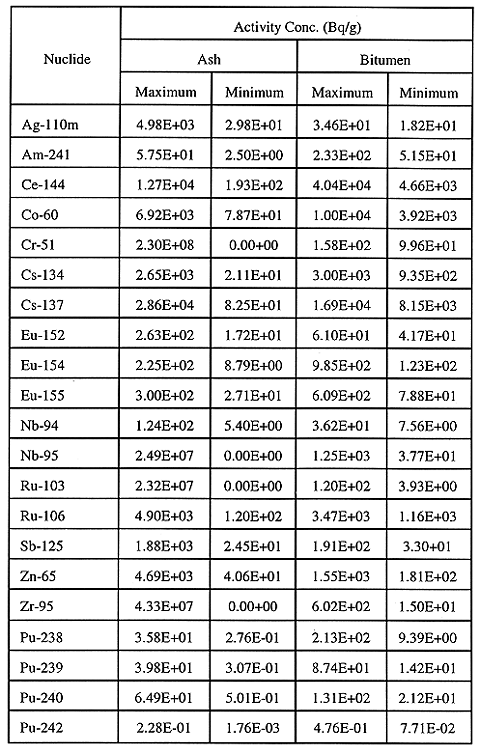
From homogeneity testing of ash samples, it was observed that the variation
in radionuclide activity concentrations among five samples from the same drum
was typically 25 to 30% (one standard deviation) while the radionuclide activity
concentrations in Table III vary from two to three orders of magnitude. Similar
variations were observed for liquid feed samples. The largest uncertainty in the
estimate of the IRUS radionuclide inventories therefore, came from the variation
in activity concentrations found in the waste, with little variation contributed
from the sampling and analysis. The total volumes of ash and bitumenized liquid
feed expected to be disposed of in the first IRUS unit (IRUS volume of 1912 m3)
were 30 m3 and 143 m3 respectively. The inventories of
the four plutonium -emitting
radionuclides in these volumes of wastes have been summarized in Table IV.
Because the -spectrometer
could not resolve 239Pu from 240Pu, a theoretical
activity ratio of 0.613 was derived for 239Pu to 240Pu
from published tables (7) of fission and activation product activity
concentrations as a function of burnup for natural uranium fuel. A burnup of one
year (800 GJ/kg uranium) with a decay period of 5 years was assumed for the
calculation.
Table IV Plutonium Inventories (Bq) in 30 m3 of Ash and
142 m3 of Bitumenized Liquid Waste

Because of the large volume of wastes handled routinely by waste management
operations, it is not possible to sample and analyze each package prior to
emplacement in a disposal facility. Therefore it is desirable to be able to
estimate inventories of long-lived radionuclides in packages using the activity
concentrations of -emitting
radionuclides determined from -monitoring. To accomplish
this, correlations between -emitting and non--emitting radionuclides
must be established through a rigorous sampling and analysis program. In the
case of plutonium, the -emitting
radionuclides which were examined for possible correlation were 137Cs,
241Am and 144Ce. The correlations for these three
radionuclides with 239+240Pu in the two waste streams are presented
in Figs. 3 to 8. The correlation between 241Am and plutonium appears
to be the best, however, due to the low energy of the 241Am -ray (59.5 keV), it is not
the best candidate for large packages due to attenuation. For large packages,
144Ce appears to be the next best candidate which can be used to
predict the plutonium activity concentration within a factor of 3 to 5 in the
two waste streams. Although the correlation is far from ideal, it may be
acceptable for those cases where the predicted plutonium activity concentration
is well below the activity concentration limit for the disposal facility.
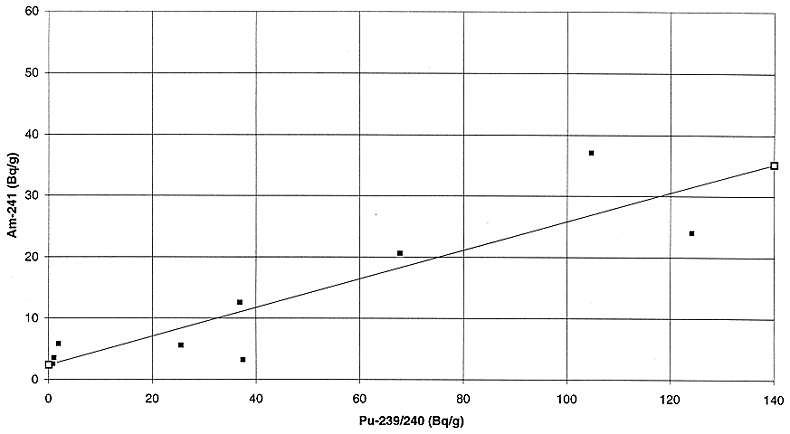
Fig. 3. Variation of plutonium
activity with Americium activity in incinerator ash.
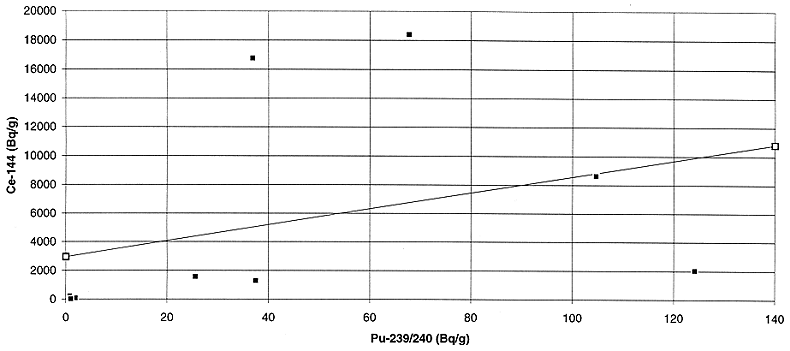
Fig. 4. Variation of plutonium
activity with cerium activity in incinerator ash.
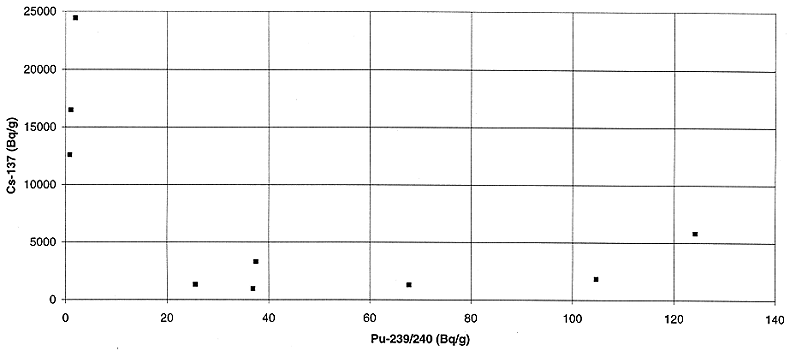
Fig. 5. Variation of plutonium
activity with cesium activity in incinerator ash.
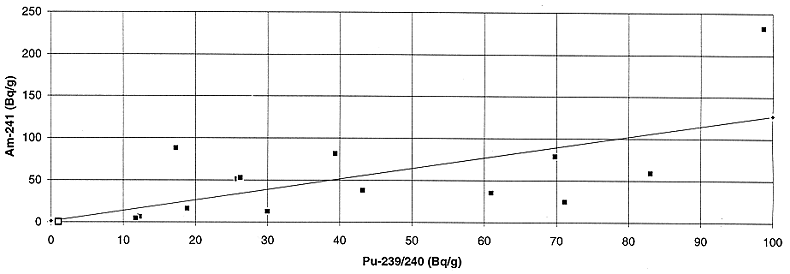
Fig. 6. Variation of plutonium
activity with americium activity in the liquid feed to the bituminerizer.
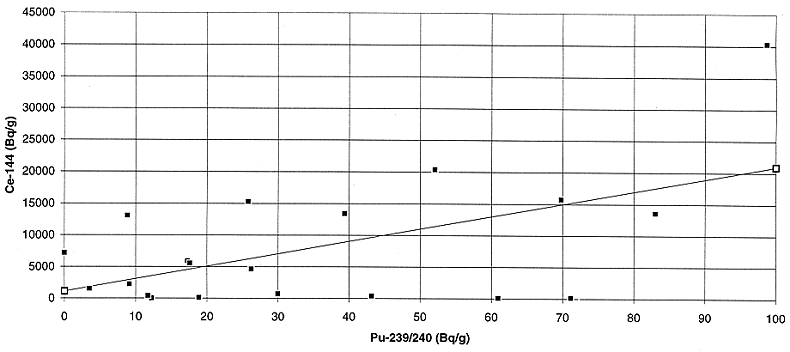
Fig. 7. Variation of plutonium
activity with cerium activity in the liquid feed to the bituminizer.
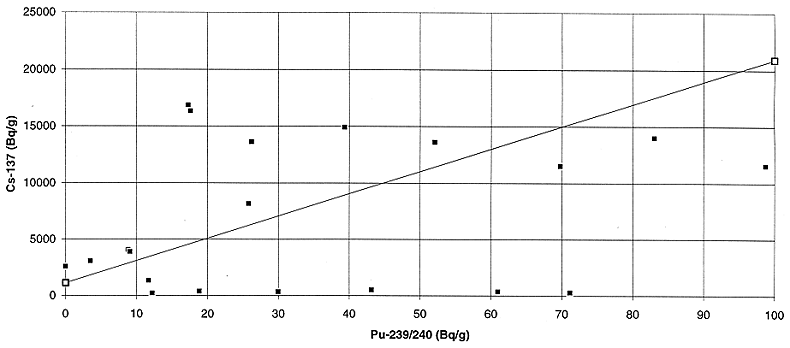
Fig. 8. Variation of plutonium
activity with cesium activity in the liquid feed to the bituminizer.
CONCLUSIONS
A method has been developed for the determination of plutonium in two
radioactive waste streams. The method has limits of detection ranging from 0.01
to 0.001 Bq/g 239+240Pu per sample in solids. The method is capable
of measuring plutonium in the presence of large amounts of uranium and is
applicable to the analysis of a variety of sample types such as soils, sludges
and fuel. It has been used to establish correlation factors with 241Am
and 144Ce which enables plutonium activity to be estimated non-
destructively through the use of -ray spectrometry to
within a factor of 3 to 5 in two waste streams. It has the added advantage that
it can be easily adapted for use with
/-discriminating liquid
scintillation counting for screening purposes.
REFERENCES
- K.I. BURNS, W.J. EDWARDS, G.W. CSULLOG, D.W. EVERALL, P.K. LEESON, and J.E.
CHEYNE, "Recent Developments in Waste Characterization at Chalk River
Nuclear Laboratories," Proceedings of the Symposium on Waste Management,
Feb.26 - Mar. 2, 1989, Waste Management '89, Volume II, pp.351-358.
- DAZHU YANG, YONGJUN ZHU, and S. MOBIUS, "Rapid Method for Alpha
Counting with Extractive Scintillator and Pulse Shape Analysis," J. of
Radioanalytical Chemistry, Articles, Vol. 147, No. 1, pp.177-189 (1991).
- J.C. WHITE, and W.J. ROSS, "Separations by Solvent Extraction with
Tri-n-octylphosphine Oxide," U.S. National Academy of Sciences, Nuclear
Science Series, NAS-NS 3102 (1961).
- DAZHU YANG, YONGJUN ZHU, and RONGZHOU JIAO, "Determination of Np, Pu
and Am in High Level Radioactive Waste with Extraction Liquid Scintillation
Counting", the 5th International Seminar for Liquid
Scintillation Counting, Proceedings of the 3rd Low Level Counting
Conference using Liquid Scintillation Analysis, pp 127-143 (1992).
- G.H. COLEMAN, "The Radiochemistry of Plutonium, U.S. National Academy
of Sciences," Nuclear Science Series, NAS-NS 3058, (1965).
- E.P. HOROWITZ, M.L. DIETZ, D.M. NELSON, J.J. LAROSA, and W.D. FAIRMAN, "Concentration
and Separation of Actinides from Urine Using a Supported Bifunctional
Organophosphorus Extractant," Anal. Chim. Acta, V238, N2, pp. 263-271
(1990).
- L.J. CLEGG, and J.R. COADY, "Radioactive Decay Properties of CANDU
Fuel, Volume 1. The Natural Uranium Fuel Cycle, Part 2: Irradiated Fuel,"
Atomic Energy of Canada Ltd. Report AECL-4436/1, (1997).