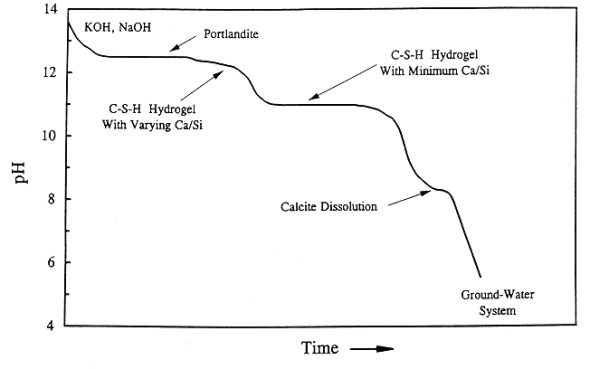
Fig. 1. Schematic diagram
illustrating the change of pore fluid pH resulting from the progressive aqueous
dissolution of cement.
A.C. Campbell
U.S. Nuclear Regulatory Commission
Washington, D.C. 20555
K.M. Krupka
Pacific Northwest National Laboratory
Richland, Washington 99352
ABSTRACT
A number of chemical considerations may be important for assessing the potential release of radionuclides from a low-level waste (LLW) disposal facility. One concern is to develop an understanding of cement/water chemistry and the chemical conditions that may occur inside the disposal units or overpack containers (e.g., pH, ionic strength, oxidation/reduction potential, chemical speciation). These conditions in turn affect solubilities and sorption properties of radionuclides. Another concern is to develop an understanding of how the chemical state of the system may evolve over time, which could affect releases of long-lived radionuclides (e.g., changes from reducing to oxidizing conditions or from high pH to lower pH). A third concern is to understand both the strengths and the weaknesses of geochemical reaction models and calculated solubility limits (e.g., thermodynamic data limitations, the role of sorptive processes, and the differences between model results and laboratory data). Consideration may also need to be given to the presence of reducing agents such as iron and organic matter or the presence of complexing agents that may enhance the mobility of radionuclides. Finally, because sorptive processes will attenuate the concentrations of most radionuclides, suitable ranges and distributions of retardation coefficients for certain radionuclides under different conceptual models need to be considered in a performance assessment.
INTRODUCTION
A key element of performance assessment (PA) modeling of LLW disposal facilities is the source term release model, which involves a large number of variables and significant uncertainty that may affect the calculated performance. The chemical environment within a LLW disposal unit will have a profound impact on the potential release of radionuclides from both waste forms and containers and the transport of radionuclides out of a disposal unit. Most States and Compacts are developing concrete vault disposal facilities in contrast to the trench disposal facilities used in the past. A concrete vault disposal unit, which is likely to contain large amounts of calcium hydroxide and calcium silicate mineral phases in various components of the system (e.g., cement waste forms, concrete over packs, grout backfill, and the structure itself), will have a strong buffering effect on the overall chemical state of the system. This, in turn, will affect parameters that control the mobility of specific radionuclides. Thus, certain assumptions about the applicability of specific ranges for solubility limits and/or retardation coefficients for key radionuclides may be appropriate in a PA for such a system. An important issue is to develop an understanding of the chemical environment that may occur in the disposal units or in the waste containers to make defendable assumptions with respect to solubility limits and sorption (Kd) or retardation (Rd) coefficients for the disposal system materials. Since performance assessment is concerned with demonstrating compliance with applicable standards, uncertainties inherent in analyzing the source term need to be considered in terms of those factors that most critically affect the ability of the disposal system to meet the performance objectives. In the near future the Nuclear Regulatory Commission will publish NUREG/CR-6377, "Effects on Radionuclide Concentrations by Cement/Ground-Water Interactions in Support of Performance Assessment of Low-level Radioactive Waste Disposal Facilities," which discusses issues and concerns and describes a foundation of information and basic approaches that can be considered in developing a PA (1). Our paper focuses on some of the issues and modeling discussed in this NUREG/CR document.
BACKGROUND
Geochemical characterization of a LLW site has been traditionally considered to be an essential component of site characterization as set forth in the "Standard Review Plan (SRP) for the Review of a License Application for a Low-Level Radioactive Waste Disposal Facility," NUREG-1200 (2). The evaluation of geochemical characteristics (SRP 2.6) includes: water chemistry, geochemistry of soil and rock units, and geochemical modeling. In addition to characterizing the natural site, geochemical information is used in evaluations of the types, kinds, and quantities of waste (SRP 6.1.1); radionuclide transfer to humans (SRP 6.1.5); intruder protection (SRP 6.2); and long term stability (SRP 6.3). In some cases, actual laboratory measurements of site and facility specific Kds are used directly in a PA. However, natural site characteristics typically display a high degree of variability and, in most cases, it is not clear how much information is needed to characterize this variance. Thus, in general, natural site geochemical information is not used directly in a PA, but is used to develop an appropriate set of "conservative" retardation coefficients. Sometimes a default set of Kd values is assumed by the analyst without consideration of the chemical conditions at the site or within the disposal system itself.
One important area in defining the chemical state of a LLW disposal system is to evaluate the chemistry of the disposal system as it is affected by the presence of cementitious components. This has not been emphasized for commercial LLW disposal in the U.S. until recently (3). Models for potential releases from a LLW disposal facility that are based on existing disposal trenches at Maxey Flats and West Valley (4) may not be appropriate for concrete vault systems because of the large differences in the chemical conditions. For example, these two trench disposal systems exhibit strongly anoxic conditions, near neutral pH, and the precipitation of carbonates and sulfides (5,6), whereas earth mounded concrete vault systems may exhibit high pH and more oxidizing conditions (1).
CEMENT/WATER CHEMISTRY
The evolution of pore water chemistry as cement degrades has been studied extensively (7, 8, 9,10). The pH of leachate solutions is one of the most important parameters in determining radionuclide solubility and sorption (1). Hence, understanding the factors that control the pH of the system and how it evolves with time is an important factor to be considered in a PA. The main components of hydrated cement are as follows: 40-50 wt% calcium silicate hydrogel (CSH); 20-25 wt% portlandite [Ca(OH)2]; 10-20 wt% ettringite [Ca6Al2O6(SO4)3], monosulfate [Ca4Al2O6SO4], and ferric phases; 10-20 wt% pore fluids; and 0-5 wt% NaOH, KOH, and Mg(OH)2. The general change in pH with time is shown graphically in Fig. 1. Some workers use the number of water "exchange cycles" (one exchange cycle is a volume of water equal to the cement pore volume) to characterize the change in pH with progressive leaching (8). Initially the leaching of potassium hydroxide (KOH) and sodium hydroxide (NaOH), buffers the pH above 13 for a relatively short-time period, (i.e., 1-100 exchange cycles). Next, leaching of portlandite and CSH, which together constitute about 60 - 75 wt% of cement, buffer the pH near 12.5 and 10.5, respectively. This buffering effect can last for relatively long-time periods, (i.e., 100 to 1,000 exchange cycle for portlandite buffering and several 1,000 exchange cycles for CSH buffering) (8). As the system is progressively leached of its constituents, the leachate solution pH approaches that of the water percolating into the system. The time period over which this evolution occurs is dependent not only on the volume of percolating water, but also on its chemical composition and the composition of cementitious material being leached (11). Preferential water flow paths through a facility may experience an accelerated evolution of pH relative to portions of the facility with lower water fluxes (e.g., within the interiors of intact concrete over packs). These factors need to be considered in a PA that takes credit for the cement buffering effect.
Fig. 1. Schematic diagram
illustrating the change of pore fluid pH resulting from the progressive aqueous
dissolution of cement.
SOLUBILITY LIMIT APPROACH
A number of geochemical modeling codes exist that are suitable for the purposes of supporting performance assessment modeling. These codes include MINTEQA2, EQ3/6, PHREEQE, GEOCHEM, MINEQL, WATEQ, and others (1). Typically these codes contain submodels that calculate the following: aqueous speciation and complexation, oxidation and reduction reactions, the degree of saturation of solid phases, the solubility of gas phases, the precipitation and dissolution of solid phases, and adsorption. In general, the codes are sufficiently complex that analyses are done independent of the PA and the results are abstracted for use in a PA Code. The codes are not generally designed to provide uncertainty ranges and distributions for direct use in a PA. In addition, thermodynamic databases used by most codes do not usually contain uncertainty values. The results, however, can provide ranges of radionuclide solubilities that are conservative maximum concentration limits. In order to further constrain the calculated solubilities and to provide more realistic ranges and distributions of radionuclide solubilities to be used in a PA, the model results need to be combined with other information such as published solubility studies and laboratory and field data.
The modeling approach can be demonstrated using calculations of uranium solubility with the MINTEQA2 geochemical reaction computer code (12). Because uranium (U) has a low specific activity, a curie of 238U corresponds to relatively large mass of the element (about 3 metric tons). Hence it is reasonable to assume that the release of U will be controlled by solubility rather than by sorption. Thermodynamic modeling provides no information on kinetic effects (i.e. the rate of dissolution). Slow kinetics will result in U concentrations below the thermodynamic equilibrium concentration and thus it is generally conservative to assume the latter. Two end member environments were considered. One is a cement-dominated system with a pH of 12.5 and an Eh of 200 millivolts (mV). This environment would represent an early stage of the disposal facility lifetime with relatively low fluxes of water percolating through the facility and where the dissolution of portlandite and CSH dominate the cement chemistry (Fig. 1). The second end member is a "ground-water" dominated system, with pH of 5.8 and Eh of 500 mV. These values are averages based on the NRC LLW PA Test Case (1). These would correspond to a system in an advanced state of degradation at this site with high water fluxes percolating through the degraded concrete vaults and where the dissolution of cement components has run to completion (Fig. 1). For the purpose of modeling, the pH and Eh are assumed to vary linearly between the two end members.
The modeling results show that dissolved uranium exists primarily in the +6 valence state. At high pH the speciation is dominated by hydroxyl complexes, whereas at the lower pH the speciation is a complicated mixture of carbonate, hydroxyl, and phosphate complexes. Two solubility controlling phases were considered, schoepite [UO2(OH)2*H20] and uranophane [Ca(H3O)2(UO2)2(SiO4)2*3H2O]. Schoepite is known to precipitate readily in low-temperature aqueous systems at laboratory time scales and result in high concentrations of dissolved uranium (13). In natural low-temperature aqueous systems, the presence of alkali and/or alkaline earth ions at high pH conditions would result in the precipitation of alkali/alkaline earth uranium compounds that would control the solubility of uranium to concentrations lower than those resulting from equilibrium with schoepite. Uranophane and other calcium uranyl compounds are known to exist in uranium-loaded C-S-H mixtures (14) and thus may be realistic solubility controls for dissolved uranium. Calculation of the solubility of calcium uranyl compounds, however, may be more susceptible to uncertainties in conceptual models and available thermodynamic data.
The calculated concentrations of dissolved uranium in the leachate are plotted as a function of pH in Fig.2. The thick solid curve represents uranophane as the solubility control and the dotted curve represents schoepite as the solubility control. Filled circles represent values estimated from experimentally determined concentrations of dissolved uranium from the equilibration of cement-equilibrated waters that were over saturated with dissolved uranium (15). The thin dotted horizontal line shows the initial concentration of total dissolved uranium used in these laboratory experiments. Results of the solubility calculations bracket the experimental data. The uranium concentrations measured at pH values of 5 and 8 are approximately equal to the concentrations used to start the over saturation experiments, and thus may not indicate solubility limited conditions in the experiments. The uranium concentrations modeled here using the solubility of schoepite are several orders of magnitude greater than the experimental values. The solubility of uranophane, on the other hand, is in good agreement with the experimental values for pH values greater than 10.5, and significantly underestimates the concentrations at lower pH values. Previous modeling studies also showed that uranium concentrations based on equilibrium with schoepite overestimate uranium concentrations relative to observed values in the high pH range (15).
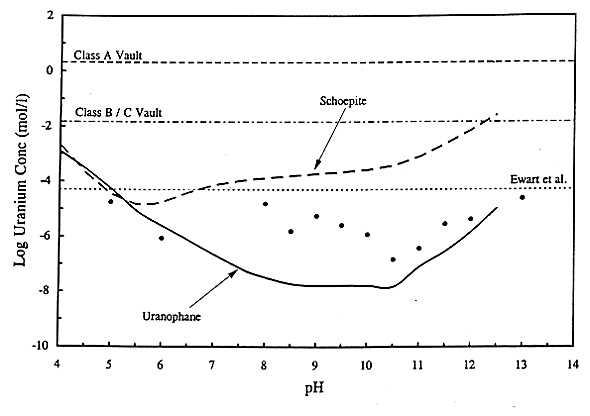
Fig. 2. Calculated
solubility limits for dissolved uranium.
In addition to pH and Eh, the calculation of solubility limited concentrations for dissolved uranium are sensitive to several other environmental parameters considered in the conceptual model. As noted from the dominant aqueous species listed above, carbonate complexation of UVI has a major effect on the maximum solubility limits calculated for dissolved uranium. Moreover, given the composition of uranophane, the concentrations of dissolved calcium and silica are additional factors affecting the calculated uranium concentrations that are based on equilibrium with uranophane.
The uranium concentrations calculated using a rinse release model from the NRC LLW PA Test Case are shown as thin dashed and dashed-dotted horizontal lines in Fig. 2. This model assumes quantitative transfer of the U inventory in a vault (prior to application of solubility limits or sorption coefficients) into water volume fluxes of 1,000 and 100 ft3/yr for 238U in ClassA and B/C vaults, respectively (1). These values were selected for plotting purposes, because they bound the highest possible uranium concentrations. These two values are very extreme relative to any reasonable estimate of release because they do not take into account physical, kinetic, or thermodynamic controls on the dissolution of U. In addition, these water flux values represent the low end of the ranges calculated in NRC's Test Case (1). The solubility limits calculated for uranium thus provide a more accurate constraint on the upper concentration limits for uranium, which can be used in a PA model.
SORPTION LIMIT APPROACH
There are a number of issues that need to be considered in developing a range of sorption values to be used in a PA for a cement-buffered LLW disposal system. These issues include the following: the relevancy of Kd values for crushed cement samples, differences in Kd values based on cement type, the difference between cement and concrete, and the long-term evolution of cement solid phases and pore waters.
Most of the Kd values measured for cement-based disposal systems have been done on crushed cement samples that have been desegregated (16,17,18,19). The process of desegregation should expose surfaces similar to those that would be expected to occur under the conditions of the disposal system as it ages. In addition, there are a limited number of studies on the effects of cement particle size on the measured Kd value. These studies generally show no significant effects of this nature (16, 18). In some cases, Kd values have been derived from monolithic samples (17,18). Based upon this information, the use of Kd values derived from experiments using crushed cement samples should be applicable for PA calculations for a cement-based disposal system (19).
Another significant issue in developing Kd values for cement-based systems is the effect that different cement types have on sorption. Additives, such as fly ash, blast furnace slags (BFS), clays and zeolites have been used in cement waste forms to better sequester the radionuclides and other contaminants (1). The evolution of pore waters for these different types of cements show generally similar chemical evolution effects, which are well defined (19). The evolution of the solid phases in these different cements are also well documented (19). One major influence on sorption is that the reduction/oxidation (Redox) potential of the cement has a significant influence on the potential for radionuclide sorption (7). For example, the addition of BFS to a cement formulation will create a reducing environment within the cement because the reduced sulfur contained in the BFS is oxidized in the degradation process (20).
Another issue is the difference between cement and concrete. Concrete is a mixture of cement, coarse aggregates and additives. The latter two components can affect the sorption properties of the concrete. Most radionuclides appear to favor chemical association with the fine-grained cement and hydration products (19). A few radionuclides show differences of adsorption on cement paste compared to concrete. For example, Cesium shows high adsorption onto the primary minerals used in aggregate (1).
The long-term evolution of pH in cement pore fluids can be conveniently divided into three different environments depending on the cement phases that is acting as the buffering agent (19). Environment I corresponds to the initial high pH, high ionic strength environment (pH>12.5) created by the leaching of NaOH and KOH from the cement matrix. Environment II corresponds to the time period when the dissolution of portlandite buffers the pH of the system to about 12.5. Environment III corresponds to the time period when the solubility of CSH phases buffers the pH to values between 12.5 and 10.5.
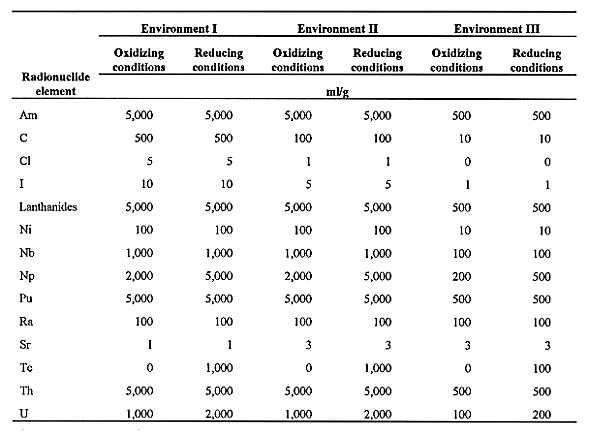
We have developed a data base of Kd values for these different cement environments that also take into account whether the environment is reducing or oxidizing (see Table I). Place Table I here. The example is illustrative of the approach that one could take in developing a basis set of Kd values for a specific site and facility design. These numbers should not be taken as a default set of values that are valid or necessarily conservative for any and all cement-based LLW systems. It is always the responsibility of the developer to provide the regulator with the rationale and basis for a particular modeling approach and to be able to defend and justify the data used in the model. In addition, for the purposes of a PA, one may wish to develop a range of values that may more realistically represent the state of knowledge about sorption of particular radionuclides on a specific type of cement to be used in a facility. This might include information from laboratory experiments, field studies, and natural analog studies.
In developing a data base for cement-based Kd values we present them as the mean or best estimate for a fresh cement paste. If it is desirable to develop a range of values for PA, then the literature cited in the NUREG/CR-6377 will provide a good starting point. In general, studies of sorption on soils have indicated that Kd values are log-normally distributed. It may be reasonable to assume that the Kd values for a cement-based system also exhibit a log-normal distribution. In addition, the preferred cement values are differentiated between oxidizing and reducing conditions. The selected values are presented in Table I. We have adopted the convention of Bradbury and Sarott (19) by assuming that in environment III (pH<12.5) the Kd value for each radionuclide is 1/10th of its value in environment II (pH=12.5). Note that some radionuclides (e.g., Tc, Np, and U) show increased sorption in a reducing environment. Also note that Sr shows increasing sorption as the cement degrades. The presence of high ionic strengths and large amounts of Ca in cement pore fluids suppress the sorption of Sr due to competition for similar sites by the alkaline earth elements and the alkali metals (1). Also note that the "sorption" of C is in fact due to isotopic exchange of radioactive carbon with stable carbon contained in carbonate phases which are stable in the high pH environment. These phases begin to dissolve as the cement degrades and the pH decreases, which results in less uptake of radiocarbon.
Table I. Preferred distribution ratio (Kd) values (ml/g)
for selected radionuclide elements for cement/concrete Environments I-III
CONCLUSIONS
This paper has discussed issues and concerns for the application of geochemical modeling and data in a performance assessment of a cement-based LLW disposal facility. There is a significant information base available in the literature for cementitious systems. These include computer modeling techniques for cement hydration pore fluids, laboratory studies, and natural analog studies (which have not been discussed in any detail in this paper). Geochemical modeling of concentration limits has demonstrated the importance of solubility modeling, the importance of selecting appropriate controlling solid phases, the important role of pH and Eh, and the importance of aqueous complexation by anionic ligands (e.g., CO32-). This paper has also demonstrated that a significant data base of Kd values exist for radionuclides in cementitious systems. The use of these data bases should provide more accurate prediction of contaminant transport from the source term in a performance assessment of a LLW disposal system.
REFERENCES