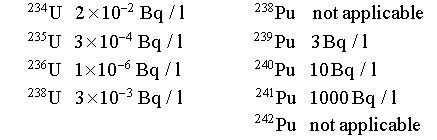
Peter De Regge , Frank Vanderlinden , Renaat Boden , Freddy
Verrezen
Studiecentrum voor Kernenergie SCK•CEN
Boeretang 200,
Mol, B-2400 Belgium
Robert Gens
Nationale Instelling voor Radioactief Afval
en Splijststoffen NIRAS/ONDRAF
Madouplein 1 b 25, Brussel, B-1210 Belgium
ABSTRACT
Safety studies related to the disposal of low- and intermediate level wastes indicate the importance for the long term risk of a series of isotopes which are not currently measurable in the waste packages because of their low content, their low specific activity or the particular characteristics of their radiation. Those isotopes are produced by activation reactions in the reactor materials (3H, 14C, 59Ni, 63Ni, 94Nb) or by fission and transmutation reactions in the nuclear fuel (90Sr, 99Tc, 129I, 135Cs, 234U, 235U, 236U, 238U, 238Pu, 239Pu, 240Pu, 241Am, 244Cm).
The inventory of those isotopes in the different PWR waste streams has been measured and possible correlations have been evaluated between those isotopes and key radionuclides, which are easily measured and representative for the occurrence of activation products (60Co) and nuclear fuel components (137Cs) in the waste streams. It has been investigated whether those correlations are valid in a general way or specific to a particular reactor and a particular waste stream as well for the Belgian reactors as in comparison with similar studies in other European countries.
The uncertainty associated with the calculated inventories for critical nuclides on the basis of the measurements of key radionuclides has been assessed. The results are based on accurate measurements of the critical nuclide content and the key radionuclides in the different waste streams, ion exchange resins (6 samples), evaporator concentrates (15 samples) and coolant filters (5 samples), for the Belgian reactors. The measurement procedures have been validated by interlaboratory comparisons applying different radiometric and mass spectrometric analytical techniques.
The measurements have been carried out in order to provide entries for a European database on disposal critical nuclides in the different countries and reactor types. The paper provides a review of the data measured for the waste streams in the Belgian nuclear power plants. Consistent and reliable correlations have been obtained for disposal critical nuclides and key nuclides in the main waste streams of Belgian nuclear power plants in routine exploitation. The measured data and the correlations have been organized into a database and are applicable to evaporator concentrates, ion exchange resins and coolant filter cartridges. The data are currently used to calibrate a mathematical model and calculation code for the prediction of disposal critical nuclide inventories in the main PWR waste streams.
INTRODUCTION
Safety studies related to the disposal of low- and intermediate level wastes indicate the importance for the long term risk of a series of isotopes which are not currently measurable in the waste packages because of their low content, their low specific activity or the particular characteristics of their radiation (1). Those isotopes are produced by activation reactions in the reactor materials (3H, 14C, 59Ni, 63Ni, 94Nb) or by fission and transmutation reactions in the nuclear fuel (90Sr, 99Tc, 129I, 135Cs, 234U, 235U, 236U, 238U, 238Pu, 239Pu, 240Pu, 241Am, 244Cm) and are further referred to as "critical isotopes".
In the framework of the fourth European Community research program on the Management and Disposal of Radioactive Waste a project entitled "Inventory and characterization of important radionuclides for safety of storage and disposal" was supported by the European Commission. The work currently reported has beneficiated from this support (2). One of the aims of this program was the checking and standardization of existing analytical methods for application to real samples from main waste streams. With the objective to increase the level of confidence in the reported results and to validate the measurement methods, the experimental program has been extended to include interlaboratory intercomparison measurements on evaporator concentrate and resin samples where each participating laboratory applied its standard measurement procedures to a common sample (3).
OBJECTIVES
A first objective is the measurement of the inventory of the critical isotopes in the different PWR waste streams and the identification of possible correlations between those isotopes and key radionuclides, which are easily measured and representative for the occurrence of activation products (60Co) and nuclear fuel components (137Cs) in the waste streams.
A second objective is to investigate whether those correlations are valid in a general way or specific to a particular reactor and a particular waste stream.
A third objective is to estimate the uncertainty associated with the calculated inventories for critical nuclides on the basis of measurements of key radionuclides.
A fourth objective is to provide calibration data for a mathematical model and calculation code developed by the Belgian utilities (Electrabel) for the prediction of disposal critical nuclide inventories in the main PWR waste streams.
The objectives are to be achieved by the accurate measurements of the critical nuclide content and the key radionuclides in the source of the waste streams, i.e. the primary coolant, and in the different waste streams for several reactors. Until now measurements have been carried out on ion exchange resins, evaporator concentrates and coolant particle filters.
ANALYTICAL METHODS
Pretreatment and dissolution of the resin samples
The results for the resin sample are all expressed per gram dry resin at a drying temperature of 105°C. Therefore the weight loss by drying the resin at 105°C during 60 hours is measured. Subsequent samples are taken from the resin after air drying at room temperature to avoid any loss of volatile isotopes. The measurement results are recalculated to the corresponding dry weight.
The resins are brought into a homogeneous solution by an oxidizing acid digestion using Fe(II) as a catalyst. Sometimes an insoluble residue amounting to a few percent of the dry resin weight is found and treated by alkaline fusion. The corresponding solution is combined with the acid digestion phase to produce an homogeneous solution used for the analyzes.
During the acid digestion off-gases are collected into a cascade of caustic soda traps to recover carbon dioxide containing the 14°C. By the use of strongly oxidizing solutions, 129I is probably transformed to iodate and has therefore not been found in the caustic soda traps.
Pretreatment and Dissolution of Evaporator Concentrates
Evaporator concentrate samples are diluted 10 times at the sampling stage and then usually consist of four identified phases: a floating or suspended pasty substance that is very difficult to filter and to dry a), a liquid phase b), boric acid crystals c) and settled out solid matter d).
All dilutions and aliquotations have been carried out by weight and their documentation allows to recalculate the isotope content of the sample or the average isotopic concentrations over all phases. The procedure however does not allow to measure individual isotope concentrations in the different phases of the sample. First a know quantity of water is added to redissolve the boric acid phase. The dissolution procedure consists of the separation of the solid and the liquid phase, air drying and weighing of the solid phase, and taking proportional parts of the solid and liquid phase to recombine a representative sample of the original concentrate. In general about 3 g of dry solids are combined with the corresponding quantity of filtrate in an acid digestion step. The remaining undissolved fraction is then filtered and treated by alkaline fusion. The resulting solution is combined with the acid digestion phase (including the filtrate) in an homogeneous representative sample solution.
Carbon -14
The sample consists of 50 ml of the combined wash solutions of dissolver off-gas. The sample solution should be alkaline or neutral. The method relies on the separation of carbon dioxide by acidolysis and the measurement of 14CO2 collected from the sample dissolution off-gases in the wash solutions. The amount of organically-bound and elemental (free) 14C is assumed to be negligible in comparison with the amount present as CO32- or HCO3-. 14C is released as CO2 by acidification of the alkaline or neutral solutions with HClO4, trapped in a flask cooled with liquid N2 and absorbed in CarboSorb® . The 14C content of CarboSorb® aliquots is measured by liquid scintillation counting according to the internal reference method (4, 5). The detection limit of 14C in the final CarboSorb® solution is about 70 Bq/l for samples containing 1 ml CarboSorb, 1 ml water and 18 ml liquid scintillation cocktail, which are counted for 1 h in a counter where the background is 30 counts per minute and the efficiency is 85 %. With a 50 ml sample and 10 ml of CarboSorb the detection limit in the initial sample (combined wash solutions) is then about 15Bq/l.
Measurement of Gamma Activities and Key Nuclides
A 25 ml aliquot is taken from the homogenized solution obtained after dissolution of the waste sample. The quantitative measurement of gamma-ray emitting nuclides is carried out according to ASTM D2459-72 (6). The following isotopes are usually observed : 57Co, 58Co, 60Co, 54Mn, 65Zn, 110mAg, 134Cs, 137Cs, and 125Sb. The counting time is 5000 to 50,000 seconds depending on the desired detection limit for the minor isotopes.
The Ge or Ge(Li) detector is calibrated according to the standard procedures cited above, using NIST standard reference sources containing 125Sb, 154Eu, and 155Eu. The reference sources have an uncertainty of 2 % at the 95% confidence level.
The detection limit in gamma-spectrometric measurements depends on the radionuclide, its gamma ray energies and branching ratios, the detector background and the overall composition of the gamma spectrum. The reference detection limit for representative spectra containing the radioisotopes cited in paragraph 6 is 10 Bq for a counting time of 50,000 s in a 25 ml sample vial. The resulting detection limit is 400 Bq/l in the original homogeneous solution. The presence of 60Co in the background radiation results in a higher detection limit around 1000Bq/l for 60Co.
Niobium-94
A 25 ml aliquot is taken from the homogenized solution obtained after dissolution of the waste sample. Niobium is separated from the interfering activities by liquid-liquid extraction. The 25ml sample is dried and redissolved in 8 M hydrochloric acid. Niobium is extracted by tributylphosphate and back-extracted with dilute hydrochloric acid. The yield of the chemical separation has been established by tracer studies and is 90 ± 5 % (1) .
The counting vial is measured by a calibrated gamma spectrometer according to the ASTM method D2459-72 (6) with a counting time of 50 000 to 150 000 s. The Ge and Ge(Li) gamma detectors have been calibrated by NIST standard reference materials containing 125Sb, 154Eu and 155Eu. The reference sources have an uncertainty of 2 % at the 95 % confidence level.
The detection limit for the 94Nb measurement in the separated fraction ranges from 8 Bq for a counting time of 50 000 seconds (resin samples) down to 0.8 Bq/l for a counting time of 150 000 seconds (evaporator concentrates). This corresponds to a detection limit of respectively 300 Bq/l and 30 Bq/l in the initial homogeneous sample solution.
Nickel-63
A sample of 20 ml is taken from the homogeneous solution obtained after dissolution of the waste sample. After the addition of nickel carrier, the nickel is then precipitated as the dimethylglyoxime complex. The complex is redissolved and the chemical separation yield is determined by Inductively Coupled Plasma Atomic Emission Spectrometry of the Ni concentration in solution. The yield ranges between 50 to 80% with an uncertainty of ± 3 % (1) on the measured value.
The efficiency of the liquid scintillation measurement is determined by an internal tracer. The 63Ni activity calibration is obtained from a reference source with an uncertainty of 10 % at the 95 % confidence level. The source is obtained from Amersham. The calibration is performed at SCK*CEN Low Level Radioactivity Measurement Laboratory using the Efficiency Tracing Technique (7).
The detection limit for the 63Ni measurement in the separated fraction is 0.2 Bq/ml for a counting time of 3600 seconds. Due to the presence of interfering isotopes and quenching the usual limit of detection is around 3 Bq/ml. This corresponds to a detection limit of 1500Bq/l in the initial homogeneous sample solution when a minimal chemical yield of 50% is obtained.
Nickel-59
The chemical separation and yield determination are carried out as for the 63Ni determination. A sample of 0.2 to 1 ml is taken from the purified nickel solution and evaporated on a disk. The counting efficiency is monitored with a 55Fe source. Counting times amount to 80,000 seconds.
The 59Ni activity calibration is obtained from a reference source of 55Fe with nearly the same X-ray energy with an uncertainty of 10 % at the 95 % confidence level. Calibration is performed at the SCK•CEN's Laboratory for Nuclear Spectrometry.
The detection limit for the 59Ni measurement in the separated fraction is 1 Bq/ml for a counting time of 80,000 seconds in a pure Ni fraction. This corresponds to a detection limit of 500Bq/l in the initial homogeneous sample solution and a chemical separation yield of 50 %.
Strontium-90
An aliquot of 25 ml is taken from the homogeneous solution obtained after dissolution of the waste sample. Strontium is separated from interfering activities on an Eichrom Sr specific column. After equilibrium is reached, 90Y is separated from the strontium fraction by precipitation as hydroxide. The yttrium precipitate is further transformed into an oxalate precipitate and mounted on a counting planchet for beta-counting.
The 90Y activity is measured with the low-level proportional counter for three counting periods of 60 minutes. Ingrowth and decay corrections are made to calculate the corresponding 90Sr activity. The chemical separation yield is determined by the addition of by 85Sr tracer. The yield is usually 100 %. The counting efficiency is determined by application of the procedure to 90Sr-90Y reference solutions. The reference solution and the tracer solution are obtained from Amersham and have an uncertainty of 2 % at the 95 % confidence level. The detection limit when using a 25 ml aliquot of the homogeneous sample solution and a counting time of 60 minutes is 1 Bq/l homogeneous solution.
Tritium
A 25 ml aliquot is taken from the homogeneous solution obtained after dissolution of the waste sample. The sample is neutralized with sodium hydroxide and introduced into a distillation flask connected to a series of traps. After evacuating the system, the first trap is cooled to -75°C by a mixture of carbon dioxide-aceton and the second one is cooled to -196°C by liquid nitrogen. The difference in water vapor pressure at the trap's temperatures distills the water from the flask into trap 1 and then into trap 2. After complete distillation of the sample, the water volume in the two traps is measured and the tritium concentration is measured in the second trap by liquid scintillation counting.
The liquid scintillation counter is calibrated by measuring a series of sealed tritium standards traceable to NIST reference material 4947. The standards have an uncertainty of 1 % at the 95 % confidence level. The detection limit for the tritium measurement in the separated fraction is about 0.03 Bq/ml for a 1ml sample counted for 1 h in a counter with a background of 20 counts per minute and an efficiency of 30 %. This corresponds to a detection limit of about 30Bq/l in the initial homogeneous sample solution.
Cesium-135
An appropriate sample is taken from the homogeneous solution or from the supernatant containing at least 1 nanogram of 137Cs or 3000 Bq. The sample size is determined on the basis of the 137Cs concentration obtained by gamma spectrometry. The 135Cs content is measured by mass-spectrometry using a thermal ionization mass-spectrometer.
Cesium is separated by extraction with Na-tetraphenylborate in amylacetate, followed by back-extraction in nitric acid. From the back-extracted Cs about 1 nanogram is loaded onto the side filaments of a triple-filament source bead and analyzed in the mass-spectrometer. The mass-spectrum is monitored and the Cs masses located by the 133Cs-isotope. All isotopes of Cs (133, 134, 135, 136, 137) are then measured relative to mass 137. Supplementary measurements of the ratio 135Cs/137Cs are made. The chemical yield is determined using the internal tracer isotopes 137Cs and 134Cs measured by gamma spectrometry.
The mass spectrometer is calibrated using different elements of varying masses : Na=23, K=39, Rb=87 and 87, Ba=136, 137 and 138, Re=185 and 187, U=235 and 238. Calibration of Cs cannot be done directly because natural Cesium is mono-isotopic. The isotope 137Cs is used as "internal" calibration.
The detection limit for the 137Cs determination in the separated fraction is 1/100 of the Cs quantity on the filament (about 1 nanogram of natural 133Cs) ; this corresponds to 10 picogram or 30 Bq 137Cs. The detection limit for the 135Cs determination is 1/10 of the mass fraction of 137Cs, which corresponds to 1 picogram or 4 x 10-5 Bq of 135Cs.
Uranium andPplutonium Isotopes
U isotopes are measured by mass-spectrometric isotopic dilution. A sample of 50 to 75 ml is spiked with about 20 ng 233U (± 7 Bq alpha) and, in the case of a mixed spike, about 0.9 ng 242Pu (± 0.14 Bq alpha) and homogenized for isotopic exchange. The sample is evaporated to dryness and redissolved in 50 ml HCl 9 M. The U and Pu spiked sample is loaded on an anion exchange column in 9 M HCl, the impurities are eluted with 8MHNO3, followed by elution of U and Pu with 0.35MHNO3.
After evaporation and redissolution an amount of solution containing 1 ng of uranium is loaded onto the side filaments of a triple-filament source bead and mass-analyzed. The mass spectrum is monitored and mass 233 is located by the 233U isotope. The other isotopes (mass 234, 235, 236 and 238) are then measured relative to 233U.
In a similar way 0.1 ng of plutonium is loaded onto the side filaments of a triple-filament bead and mass-analyzed. The mass spectrum is monitored and mass 242 located by the 242Pu isotope. The other isotopes (mass 239, 240 and 241) are then measured relative to 242Pu. The amount of 238Pu and 241Pu are usually too small to be measured by this technique.
The mass-spectrometer is calibrated with NIST or EC-NRM uranium and plutonium standards with known isotopic composition. The absolute quantities of individual uranium isotopes are obtained from their ratios with respect to 233U. The 233U concentration in the spike solution is usually known with an accuracy better than 0.2 %.The absolute quantities of individual plutonium isotopes are obtained from their ratios with respect to 242Pu. The 242Pu concentration in the spike solution is usually known with an accuracy better than 0.5 %.
The detection limit is dictated by the uncertainty on the uranium isotopic concentrations in the reagent blank and in the 233U spike. At present a reagent blank around 13 ng is observed with an assumed uncertainty of 100 %. The isotopic composition of the blank is nearly natural uranium. The 233U spike is highly enriched with a 234U concentration of 0.28 %.
Similarly the detection limit is dictated by the uncertainty on the plutonium isotopic concentrations in the reagent blank and in the 242Pu spike. At present a reagent blank around 0.06ng is observed with an assumed uncertainty of 0.02 ng. The 242Pu spike is highly enriched and has a 239Pu concentration of 0.08 % and a 240Pu concentration of 0.01 %.
The following detection limits are then deduced for the different uranium and plutonium isotopes when a 50 ml aliquot sample of the homogeneous solution is used for the analysis :
RESULTS
Individual measurements vary over a wide range as a result of specific reactor exploitation schedules and maintenance interventions during shutdown and refuelling. Therefor the results are presented as activity ratios with respect to a selected key nuclide. It is anticipated that the activity ratios will show a constant scaling factor or at least display a reproducible correlation. The results obtained for the power plant sites of CN Tihange (3 units) and KC Doel (4 units) are shown in Table I. Because individual results for specific ratios may vary to a considerable extent it was felt more appropriate to characterize the scaling factor as a geometric mean and to calculate the corresponding variance from their relative distribution around this geometric mean. This presentation provides a straightforward way to estimate the uncertainty associated with the use of the scaling factors when applied to individual waste streams and to judge the significance of differences observed between the individual waste streams, waste batches and NPP sites.
TABLE I Scaling Factors for Waste Streams at Belgian NPP Sites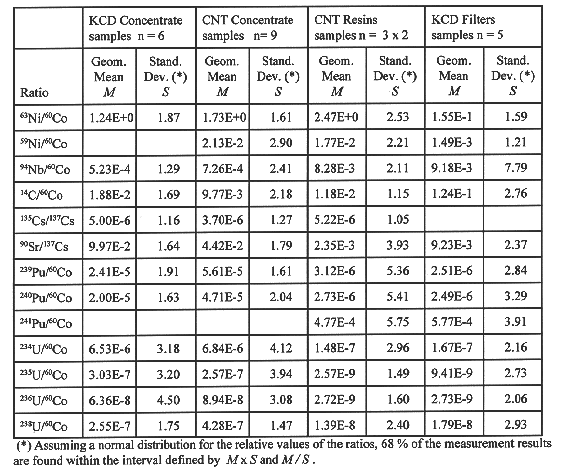
Evaporator concentrates
From Table I it can be seen that the critical isotope inventories in the evaporator concentrates for both NPP sites can be estimated within a factor of two (68 % confidence interval) except the minor uranium isotopes and 59Ni. The mean scaling factor for any individual isotopic ratio also agrees within a factor of two between the two NPP sites.
Ion exchange resins
The results for ion exchange resins were obtained on duplicate samples for three different batches processed by CN Tihange. The six results were pooled in Table I and include both sample and batch variability. Detailed examination however shows that the between batch variance dominates over sampling variance for most isotopes. Isotopic ratios of the same element are quite reproducible and for example 59Ni/63Ni=0.0073±0.0013 and 135Cs/137Cs=5.2E-6±0.3E-6, although individual concentrations per batch vary over a factor of 20 and also the scaling factor for 14C is quite reproducible. The between batch variation of the scaling factors for other isotopes are very probably significant but the causes for this variation are not well understood and further investigations are needed before those scaling factors can be applied for resin waste characterization with a reasonable uncertainty margin.
For the nickel isotopes, for 14C and for 135Cs the mean scaling factor is in the same range as for the evaporator concentrates. Considerable differences however are observed for the scaling factor of 94Nb, which is about 10 times larger, and for 90Sr and the actinide isotopes which are about 20 to 30 times smaller.
Coolant System Particle Filters
The variability on the results for the particle filters is somewhat less than is observed for the ion exchange resins, except for 94Nb. The scaling factors for the filters and the resins are quite similar for the actinides. The scaling factor for 90Sr is four times higher than for the resins but still one order of magnitude smaller than in the evaporator concentrate. The scaling factors for the nickel isotopes and 14C are respectively 10 times lower and 10 times higher than their corresponding values in the resins and in the evaporator concentrates. When considering the expected range of the scaling factors as derived from their standard deviations, the observed differences are statistically significant and should be accounted for in the development of the mathematical model.
VALIDATION OF THE RESULTS
The opportunity of an interlaboratory measurement evaluation organized within Europe in the framework of the fourth European Community research program on the Management and Disposal of Radioactive Waste was taken to assess the uncertainty components induced by the analytical methods and to validate the methods by comparison of the results with results obtained by different laboratories and methods on a common homogeneous sample.
For this purpose an ion exchange resin sample was selected. Four subsamples were taken and dissolved individually according to the standard procedure in order to produce four homogeneous solutions. Radiochemical separations and measurements were carried out in duplicate on each solution producing eight individual measurement results. In a first step the results were pooled in order to assess the combined variance due to subsampling, dissolution and radiochemical analysis method. The observed within-laboratory standard deviations (8) are shown in Table II.
TABLE II Validation of Radioanalytical Methods on Dry Resin Samples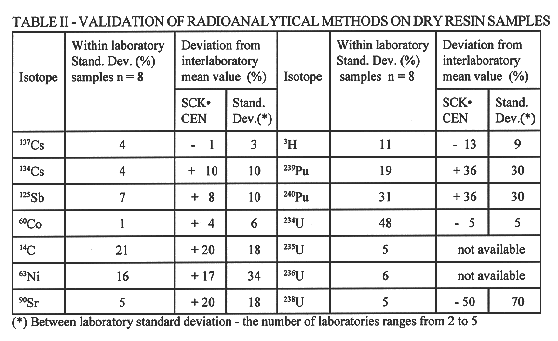
The interlaboratory evaluation also provides an average value for the isotopic concentrations in the resin batch and a between-laboratory standard deviation. In the absence of an independent characterization of the resin, the interlaboratory average, obtained by different measurement techniques, is considered as the best estimate of the true isotopic contents. The deviation of the SCK•CEN reported value with respect to this average value and the between-laboratory standard deviations are also shown in Table II. For most isotopes the within-and the between-laboratory standard deviation and SCK•CEN's deviation from the average are quite similar and can be used as an estimate for the analytical method's capability.
CONCLUSIONS
Radioanalytical methods have been developed for the determination of isotopic concentrations of critical nuclides 3H, 14C, 59Ni, 63Ni, 94Nb, 90Sr, 135Cs, 234U, 235U, 236U, 238U, 239Pu, 240Pu and 241Pu in NPP low level waste streams. Methods under development have still to be tested for 99Tc, 129I, 241Am, 242Cm and 244Cm. The methods have been applied to different batches of a variety of low level waste streams over a time interval of a few years in order to determine the scaling factors for the critical nuclides with respect to the key radionuclides 60Co and 137Cs.
Evaporator concentrates for both NPP sites display a quite homogeneous distribution allowing the use of average scaling factors to predict the isotopic concentrations of critical nuclides within a factor of two. The precision and accuracy of the radioanalytical methods, as obtained from an interlaboratory intercomparison, are at least ten times better and therefore there is at present no need to improve the capability of the measurement procedures for this purpose.
Significant differences between batches have been observed for the scaling factors in ion exchange resins and coolant particle filters. For most isotopes the scaling factors show also a significant difference between the different waste streams in addition to the between-batch differences. The development of a mathematical model to support the scaling factors should therefore incorporate the detailed aspects of reactor water chemistry to account for those differences. The accuracy and precision of the current results considered as satisfactory for the calibration of this model.
REFERENCES